Plecasoft Tablets


1.0 Generic name
Plecanatide Tablets 3 mg
2.0 Qualitative and quantitative composition
Each uncoated tablet contains :
Plecanatide 3 mg
3.0 Dosage form & strength
Tablet 3 mg
4.0 Clinical particulars
4.1 Therapeutic indications
Indicated in adults for the treatment of
- Chronic idiopathic constipation (CIC)
- Irritable bowel syndrome with constipation (IBS-C)
4.2 Posology and method of administration
The recommended dosage of Plecasoft for the treatment of CIC and IBS-C is 3 mg taken orally once daily. Preparation and administration instructions
- Plecasoft can be taken with or without food.
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take two doses at the same time.
- Swallow a tablet whole for each dose.
- For adult patients with swallowing difficulties, Plecasoft tablets can be crushed and administered orally either in applesauce or with water or administered with water via a nasogastric or gastric feeding tube. Mixing Plecasoft crushed tablets in other soft foods or in other liquids has not been tested.
Oral administration in applesauce
- In a clean container, crush the Plecasoft tablet to a powder and mix with 1 teaspoonful of room temperature applesauce.
- Consume the entire tablet-applesauce mixture immediately. Do not store the mixture for later use.
Oral administration in water
- Place the Plecasoft tablet in a clean cup.
- Pour approximately 30 mL of room temperature water into the cup.
- Mix by gently swirling the tablet and water mixture for at least 10 seconds. The Plecasoft tablet will fall apart in the water.
- Swallow the entire contents of the tablet water mixture immediately.
- If any portion of the tablet is left in the cup, add another 30 mL of water to the cup, swirl for at least 10 seconds, and swallow immediately.
- Do not store the tablet-water mixture for later use.
Administration with water via a nasogastric or gastric feeding tube
- Place the Plecasoft tablet in a clean cup with 30 mL of room temperature water.
- Mix by gently swirling the tablet and water mixture for at least 15 seconds. The Plecasoft tablet will fall apart in the water.
- Flush the nasogastric or gastric feeding tube with 30 mL of water using a catheter tip syringe.
- Draw up the mixture using the syringe and immediately administer via the nasogastric or gastric feeding tube. Do not reserve for future use.
- If any portion of the tablet is left in the cup, add another 30 mL of water to the cup, swirl for at least 15 seconds, and using the same syringe, administer via the nasogastric or gastric feeding tube.
- Using the same or a fresh syringe, flush the nasogastric or gastric feeding tube with at least 10 mL of water.
4.3 Contraindications
- Patients less than 6 years of age due to the risk of serious dehydration.
- Patients with known or suspected mechanical gastrointestinal obstruction.
4.4 Special warnings and precautions for use
Risk of serious dehydration in pediatric patients
Plecanatide is contraindicated in patients less than 6 years of age. The safety and effectiveness of Plecanatide in patients less than 18 years of age have not been established Due to increased intestinal expression of GC-C, patients less than 6 years of age may be more likely than patients 6 years of age and older to develop severe diarrhea and its potentially serious consequences. Avoid the use of Plecanatide in patients 6 years to less than 18 years of age. Diarrhea If severe diarrhea occurs, suspend dosing and rehydrate the patient.
4.5 Drugs interactions
Neither plecanatide nor its active metabolite inhibited the cytochrome P450 (CYP) enzymes 2C9 and 3A4, and they did not induce CYP3A4 in vitro. Plecanatide and its active metabolite were neither substrates nor inhibitors of the transporters Pglycoprotein (P-gp) or breast cancer resistance protein (BCRP) in vitro.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Pregnancy
Plecanatide and its active metabolite are negligibly absorbed systemically following oral administration and maternal use is not expected to result in fetal exposure to the drug. The available data on Plecanatide use in pregnant women are not sufficient to inform any drug associated risks for major birth defects and miscarriage. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes.
Lactation
There is no information regarding the presence of plecanatide in human milk, or its effects on milk production or the breastfed infant. Plecanatide and its active metabolite are negligibly absorbed systemically following oral administration. It is unknown whether the negligible systemic absorption of plecanatide by adults will result in a clinically relevant exposure to breastfed infants. Exposure to plecanatide in breastfed infants has the potential for serious adverse effects. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Plecanatide and any potential adverse effects on the breastfed infant from Plecanatide or from the underlying maternal condition.
Pediatric use
Plecanatide is contraindicated in pediatric patients less than 6 years of age. Avoid use of Plecanatide in patients 6 years to less than 18 years of age. The safety and effectiveness of Plecanatide in patients less than 18 years of age have not been established. Because of increased intestinal expression of GC-C, patients less than 6 years of age may be more likely than patients 6 years of age and older to develop diarrhea and its potentially serious consequences. Plecanatide is contraindicated in patients less than 6 years of age.
Geriatric use
The safety and effectiveness of Plecanatide in patients greater than 65 years of age have not been established.
4.7 Effects on ability to drive and use machines
Plecanatide has no or negligible influence on the ability to drive and use machines. During treatment with Plecanatide, dizziness has been reported as less common adverse reaction. Therefore, patients who experience dizziness should be cautious while driving or using machines.
4.8 Undesirable effects

Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.co.in/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is no specific treatment to the event of overdose. In the event of overdose, the patient should be treated symptomatically and supportive measures instituted as required.
5.0 Pharmacological properties
5.1 Mechanism of action
Plecanatide is a structural analog of human uroguanylin, and similarly to uroguanylin, plecanatide functions as a guanylate cyclase-C (GC-C) agonist. Both plecanatide and its active metabolite bind to GC-C and act locally on the luminal surface of the intestinal epithelium. Activation of GC-C results in an increase in both intracellular and extracellular concentrations of cyclic guanosine monophosphate (cGMP). Elevation of extracellular cGMP has been associated with a decrease in the activity of pain-sensing nerves in animal models of visceral pain. Elevation of intracellular cGMP stimulates secretion of chloride and bicarbonate into the intestinal lumen, mainly through activation of the cystic brosis transmembrane conductance regulator (CFTR) ion channel, resulting in increased intestinal fluid and accelerated transit.
5.2 Pharmacodynamic properties
Food effect
Subjects who received either a low-fat, low calorie (LF-LC) meal or a high fat, high calorie (HF-HC) meal reported looser stools than fasted subjects up to 24 hours after a single dose of plecanatide 9 mg (3 times the recommended dose). In clinical studies, plecanatide was administered with or without food.
5.3 Pharmacokinetic properties
Absorption
Plecanatide was minimally absorbed with negligible systemic availability following oral administration. Concentrations of plecanatide and its active metabolite in plasma were below the limit of quantitation in the majority of analyzed plasma samples after an oral plecanatide dose of 3 mg. Therefore, standard pharmacokinetic parameters such as AUC, Cmax, and half-life (t1/2) could not be calculated.
Distribution
Given that plecanatide concentrations following clinically relevant oral doses were not measurable, plecanatide is expected to be minimally distributed in tissues. Oral plecanatide was localized to the GI tract where it exerted its effects as a GC-C agonist with negligible systemic exposure. Plecanatide exhibited little to no binding to human serum albumin or human α-1-acid glycoprotein.
Metabolism
Plecanatide was metabolized in the GI tract to an active metabolite by loss of the terminal leucine moiety. Both plecanatide and the metabolite were proteolytically degraded within the intestinal lumen to smaller peptides and naturally occurring amino acids.
Excretion
Plecanatide and its active metabolite were not measurable in plasma following administration of the recommended clinical doses.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
The carcinogenic potential of plecanatide was assessed in 2-year carcinogenicity studies in mice and rats. Plecanatide was not tumorigenic in mice at oral doses up to 90 mg/kg/day or in rats at oral doses up to 100 mg/kg/day. Limited systemic exposure to plecanatide was achieved at the tested dose levels in animals, whereas no detectable exposure occurred in humans. Therefore, animal and human doses should not be compared directly for evaluating relative exposure. Plecanatide was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, in vitro mouse lymphoma mutation assay, or the in vivo mouse bone marrow micronucleus assay. Plecanatide had no effect on fertility or reproductive function in male or female mice at oral doses of up to 600 mg/kg/day.
7.0 Description
Plecasoft (plecanatide) is a guanylate cyclase-C (GC-C) agonist. Plecanatide is a 16 amino acid peptide with the following chemical name : L-Leucine, L-asparaginyl-L-α-aspartyl-L-α-glutamyl-L-cysteinyl-L-α-glutamyl-L-leucyl-L-cysteinyl-L-valyl-L-asparaginyl-L-valyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-, cyclic (4 12), (7 15)-bis (disufide).The molecular formula of plecanatide is C65H104N18O26S4 and the molecular weight is 1682 Daltons.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Alu-Alu blister strip of 10 tablets.
8.4 Storage and handling instructions
Store at a temperature below 25°C.
Keep out of reach of children.
9.0 Patient counselling information
Advise patients :
Diarrhea
To stop Plecasoft and contact their healthcare provider if they experience severe diarrhea.
Accidental ingestion
Accidental ingestion of Plecasoft in children, especially in children less than 6 years of age, may result in severe diarrhea and dehydration. Instruct patients to take steps to store Plecasoft securely and out of reach of children and to dispose of unused Plecanatide.
Administration and handling instructions
- To take Plecasoft once daily with or without food
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take two doses at the same time.
- To swallow Plecasoft tablets whole.
- If adult patients have swallowing difficulties, Plecasoft tablets can be crushed and administered orally in either applesauce or with water, or administered with water via a nasogastric or gastric feeding tube. • To keep Plecasoft in a dry place. Protect from moisture. For bottles, keep Plecasoft in the original bottle. Do not remove desiccant from the bottle. Do not subdivide or repackage. Remove and discard polyester coil after opening. Keep bottles closed tightly.
12.0 Date of issue
21 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Plecasoft is and what it is used for
- What you need to know before you take Plecasoft
- How to take Plecasoft
- Possible side effects
- How to store Plecasoft
- Contents of the pack and other information
1. What Plecasoft is and what it is used for
Plecasoft contains the active ingredient plecanatide, which works directly in the intestine to help increase fluid and support regular bowel movements. It acts like a natural substance in your body to draw water into the intestines, making stools softer and easier to pass. This makes stools softer and easier to pass. It helps relieve symptoms like hard stools, bloating, and stomach discomfort. Plecasoft is used in adults to treat long-term(chronic) constipation and constipation caused by irritable bowel syndrome (IBS-C).
2. What you need to know before you take Plecasoft
Do not take Plecasoft if you:
- Are less than 6 years of age (due to risk of serious dehydration)
- Have a known or suspected mechanical obstruction in your gut/intestine
Warnings and precautions
Talk to your doctor before taking Plecasoft if:
- You are between 6 to 18 years of age, as it is not recommended in this age group.
- You experience severe diarrhoea, in which case treatment should be stopped and you should consult your doctor.
Children and adolescents
Do not give this medicine to children under 6 years of age. Avoid use in children and adolescents aged 6 to less than 18 years.
Other medicines and Plecasoft
Plecasoft has a low likelihood of interacting with other medicines. However, always inform your doctor or pharmacist if you are taking or have recently taken any other medicines.
Pregnancy and breast-feeding
- This medicine is not absorbed significantly in the body; however, its safety in pregnancy is not fully established.
- It is unknown whether Plecanatide passes into breast milk. Speak to your doctor before using this medicine if you are pregnant or breastfeeding.
Driving and using machines
Plecasoft has no or negligible influence on the ability to drive or use machines. However, dizziness has been reported in some cases.
3. How to take Plecasoft
Dosage
The recommended dose is one tablet (3 mg) once daily, with or without food.
Administration
- Swallow the tablet whole.
- If you have trouble swallowing:
- You may crush the tablet and mix with 1 teaspoon of applesauce or 30 mL of water.
- It can also be administered via a nasogastric or gastric feeding tube.
- Always consume or administer the mixture immediately. Do not store it for later use.
- Follow your doctor’s instructions.
If you miss a dose
Skip the missed dose. Take your next dose at the regular time. Do not take two doses at the same time.
4. Possible side effects
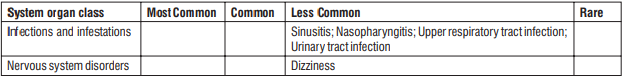
Less common:
- Nausea
- Abdominal distension
- Flatulence
- Sinusitis
- Nasopharyngitis
- Upper respiratory tract infection
- Urinary tract infection
- Dizziness
- Increased liver enzyme levels
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Plecasoft
- Store below 25°C.
- Keep in the original packaging to protect from moisture.
- Keep out of sight and reach of children.
- Do not use after the expiry date stated on the pack.
6. Contents of the pack and other information
What Plecasoft contains
- The active substance is plecanatide 3 mg per tablet.
What Plecasoft looks like and contents of the pack
- Plecasoft is available in Alu-Alu blister packs of 10 tablets.
Marketing Authorisation Holder
Zuventus Healthcare Ltd. Plot Y2, CTS No. 358/A2, Near Nahur Railway Station, Nahur (West), Mumbai - 400 078, India.
This leaflet was last revised in May 2025.
For More Information About This Product
Gutclear 200 ml Syrup
1.0 Generic name
Lactitol Monohydrate Syrup 66.67% w/v
2.0 Qualitative and quantitative composition
Each 15 ml syrup contains:
Lactitol Monohydrate USP 10 g
Benzoic Acid USP 0.0225 g
3.0 Dosage form and strength
Syrup
10 g/15ml in 100/200 ml bottles
4.0 Clinical particulars
4.1 Therapeutic indications
Gutclear® Syrup is indicated for treatment of chronic constipation and prevention of hepatic encephalopathy.
4.2 Posology and method of administration
In the treatment of constipation, the recommended dose of Gutclear® Syrup is as follows
Adults:
The recommended adult dosage of Gutclear® Syrup is 30 ml (20 grams) orally once daily, preferably with meals.
Reduce the dosage to 15 ml (10 grams) once daily for persistent loose stools.
Pediatrics: 250-400 mg/kg/day
Gutclear® Syrup is given as a single dose with the morning or evening meal, subsequently adjusted to produce one stool daily.
In the treatment of hepatic encephalopathy, Gutclear® Syrup is given in usual oral doses of 500 to 700 mg/kg daily in 3 divided doses at meal times. The dose is subsequently adjusted to produce 2 soft stools daily.
Administration Instructions
Doses should be mixed with food or liquid, and it is recommended to drink 1 to 2 glasses of liquid with the meal.
Administer other oral medications at least 2 hours before or 2 hours after Gutclear® Syrup.
4.3 Contraindications
Gutclear® Syrup is contraindicated in:
- Patients with the undiagnosed abdominal pain, colic, bleeding or vomiting
- Patients with intestinal obstruction, ileostomy, colostomy, appendicitis or diverticulitis
- Patients with galactosemia
- Patients hypersensitive to the drug or any other component of the formulation
4.4 Special warnings and precautions for use
- It is suggested that individuals taking Gutclear® Syrup have their fluid and salt (electrolyte) balance monitored regularly especially in elderly patients on long term treatment.
- Treatment with Gutclear® Syrup may cause accumulation of hydrogen in the bowel; patient should be advised to have a thorough bowel cleansing with a non-fermentable solution.
4.5 Interaction with other medicinal products and other forms of interaction
Reduced Absorption of Other Oral Medications Gutclear® Syrup may reduce the absorption of concomitantly administered oral medications. Administer oral medications at least 2 hours before or 2 hours after Gutclear® Syrup [see Dosage and Administration] Gutclear® Syrup should not be administered with other laxatives.
Caution may be exercised in using Gutclear® Syrup with drugs causing potassium loss like loop diuretics.
Gutclear® Syrup can increase digitalis toxicity.
Reduction in acidification effect can be observed if broad spectrum antibacterial agents or antacids are administered along with Gutclear® Syrup.
4.6 Pregnancy and lactation
Pregnancy
Lactitol is minimally absorbed systemically following oral administration, and it is unknown whether maternal use will result in fetal exposure to the drug. Available data from case reports on lactitol use in pregnant women are insufficient to evaluate for any drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal developmental studies, no effects on embryo-fetal development were observed with oral administration of lactitol to rats and rabbits during organogenesis at doses much higher than the maximum recommended human dosage. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the United States general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Gutclear® Syrup should be prescribed only if the potential benefits outweigh the risks to the fetus.
Animal Data
Reproduction studies have been performed in pregnant rats at oral doses of lactitol up to 2 g/kg/day (about 0.93 times the recommended daily human dose based on body surface area) and in pregnant rabbits at oral doses up to 1 g/kg/day (about 0.93 times the recommended daily human dose based on body surface area) administered during the period of organogenesis. These studies did not reveal any evidence of harm to the fetus due to lactitol.
In a pre-and postnatal development study in rats, lactitol, administered from gestation day 6 to lactation day 20, did not cause any adverse effect on pre and postnatal development up to 2 g/kg/day (about 0.93 times the recommended daily human dose based on body surface area).
Lactation
There are no data on the presence of lactitol in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Lactitol is minimally absorbed systemically following oral administration. It is unknown whether the minimal systemic absorption of lactitol by adults will result in a clinically relevant exposure to breastfed infants. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Gutclear® Syrup and any potential adverse effects on the breastfed infant from Gutclear® Syrup or from the underlying maternal condition.
No studies are available on the secretion of lactitol in breast milk.
4.7 Effects on ability to drive and use machines
Lactulose has no or negligible influence on your ability to drive safely or use machines.
Undesirable effects
The most commonly observed adverse effects with Gutclear® Syrup are abdominal cramps, distensionor flatulence during the first 10 days of treatment which are likely to disappear after continued administration. The other less frequent side effects are abdominal pain, diarrhoea, nausea and vomiting, anal pruritus, borborygmii or steatorrhea.
Reporting of suspected adverse reactions
- Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
- Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.8 Overdose
No data on overdosage of Lactitol is available. If a patient consumes large amount of Lactitol, symptoms of abdominal pain and diarrhea may appear. There may also be electrolyte imbalance. There is no specific antidote known for Lactitol over dosage. Gastric lavage may be instituted at the earliest to remove the remnant drug from the stomach. Patient should be treated symptomatically and electrolyte levels should be monitored periodically.
5.0 Pharmacological properties
5.1 Pharmacodynamic properties
Mechanism of action
Once ingested, Lactitol is neither hydrolysed nor absorbed in the small intestine. It is passed undigested to the colon and a substantial proportion of orally ingested lactitol becomes substrate for the resident colonic microflora where it is slowly fermented and is converted into short-chain fatty acids (SCFAs). The liberation of short-chain fatty acids causes a fall in pH of the right colon. The fall in pH results in the formation of an acidic media.
The reduction in intra-luminal pH increases the intra-luminal osmolality. This promotes retention of water within the bowel lumen, softening the stool and increasing the bowel volume. Hydration of the gut content and reduction in intra-luminal pH also results in shorter transit time and the establishment of laxation.
Lactitol decreases blood ammonia concentration by inhibiting both the production and the absorption of ammonia by reducing intestinal pH and shortening the residence time of intestinal contents in the intestinal tract and hence improves hepatic encephalopathy. Lactitol also favors the growth of saccharolytic (healthy) bacteria in the gut.
5.2 Pharmacokinetic Properties
Following a single oral dose of 20-gram Lactitol in healthy adult subjects under fed conditions, the mean ± SD peak serum concentration (Cmax) is 776 ± 253 ng/mL, and the mean ± SD area under the curve (AUC) is 6,019 ± 1,771 ng*hr/mL.
Absorption
Lactitol is minimally absorbed systemically following oral administration. The mean ± SD time to reach peak serum concentration (Tmax) is 3.6 ± 1.2 hours following a single oral dose of 20-gram Lactitol in healthy adult subjects under fed conditions.
Effect of Food
Cmax and AUC values increase greater than 2-fold under fasted conditions compared to fed conditions.
Elimination
The mean half-life of lactitol is 2.4 hours.
Excretion
6.0 Nonclinical properties
6.1 Preclinical safety data
A perinatal and postnatal study of lactitol, a hepatic encephalopathy drug was conducted in Sprague-Dawley rats. Female rats were given lactitol orally at dose levels of 0 (control), 0.7, 2.65 and 10 g/kg from day 17 of pregnancy to day 21 after delivery. All pregnant rats per level were allowed to deliver naturally for postnatal examination of their offspring. The high dose caused diarrhea or soft stool in dams. The high dose suppressed the body weight of dams during the perinatal period. The food consumption of dams decreased in the intermediate and high dose groups. The water consumption of dams increased in the high dose group. The high dose caused enlargement of cecum and increase of weights of cecum in dams. The drug failed to affect the delivery of dams and gestation index. However, high dose caused prolongation of gestation period. Two dams in the high dose group failed to nurse their all newborns during early lactation. The drug did not affect the number of live newborns, birth index, external appearance, body weight, viability index, weaning index, and sex ratio of weanlings. Nor did lactitol have any adverse effect on the postnatal development of the first (F1) generation offspring, such as differentiation, emotionality, motor ability, learning ability or reproductive performance. Nor did lactitol have any adverse effect on the second (F2) generation offspring. The results show that the no-effect dose levels of lactitol are 0.7 g/kg for general toxicity in mother animals, 2.65 g/kg for reproductive function in mother animals, and 10 g/kg for their offspring.
7.0 Description
Lactitol is an osmotic laxative for oral use. Lactitol is a simple monosaccharide sugar alcohol. It is a dry, free flowing, white to off-white powder, readily soluble in aqueous solutions. As shown by the structure diagrams, it is an analog of the disaccharide lactulose. Its chemical name is 4-O-β-dGalactopyranosyl-d-glucitol lactitol.

Molecular Formula C12H24O11
Molecular Weight 344.31
Gutclear® Syrup ® (lactitol) for oral solution is available in multi-dose bottles.
8.0 Pharmaceutical particulars
8.1 List of excipients
Benzoic Acid and Purified water
8.2 Incompatibilities
Gutclear® Syrup may reduce the absorption of concomitantly administered oral medications. Administer oral medications at least 2 hours before or 2 hours after Gutclear® Syrup
8.3 Shelf life
36 months
8.4 Special precautions for storage
Store below 30°C. Protect from light. Do not freeze
8.5 Nature and contents of container
100 ml or 200 ml filled in amber colour pet bottle of 200 ml with a silver pp cap with ep wad and 15 ml measuring cup. One such pack is placed along with a leaflet in a carton.
8.6 Special precautions for disposal and other handling
Any unused product or waste material should be disposed of in accordance with local requirements.
9.0 Patient Counselling Information
Persistent Loose Stools Advise patients to stop Gutclear® Syrup ® Syrup and contact their healthcare provider if they experience persistent loose stools.
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
Always take this medicine exactly as described in this leaflet or as your doctor, pharmacist or nurse has told you.
- Keep this leaflet. You may need to read it again.
- Ask your pharmacist if you need more information or advice.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet.
- You must talk to a doctor if you do not feel better or if you feel worse.
What is in this leaflet:
- What Gutclear® Syrup is and what it is used for
- What you need to know before you take Gutclear® Syrup ® Syrup
- How to take Gutclear® Syrup
- Possible side effects
- How to store Gutclear® Syrup
- Contents of the pack and other information
1. What Gutclear® Syrup is and What It is Used for
The name of this medicine is Gutclear® Syrup, which contains lactitol. It is a laxative for the treatment of chronic constipation and prevention of hepatic encephalopathy in adults, adolescents and elderly. Lactitol exerts an osmotic effect, causing the influx of water into the small intestine leading to a laxative effect in the colon.
Lactitol promotes retention of water within the bowel lumen, softening the stool and increasing the bowel volume. Hydration of the gut content and reduction in intra-luminal pH also results in shorter transit time and the establishment of laxation.
Lactitol decreases blood ammonia concentration by inhibiting both the production and the absorption of ammonia by reducing intestinal pH and shortening the residence time of intestinal contents in the intestinal tract and hence improves hepatic encephalopathy.
2. What You Need to Know Before You Take Gutclear® Syrup
Do not take Gutclear® Syrup if your doctor has told you that you have:
- Patients with the undiagnosed abdominal pain, colic, bleeding or vomiting.
- Patients with intestinal obstruction, ileostomy, colostomy, appendicitis or diverticulitis.
- Patients with galactosemia (disease which impairs the body's ability to process and produce energy from a sugar called galactose)
- Patients hypersensitive to the drug or any other component of the formulation.
Warnings and Precautions
- It is suggested that individuals taking Gutclear® Syrup have their fluid and salt (electrolyte) balance monitored regularly especially in elderly patients on long term treatment.
- Treatment with Gutclear® Syrup may cause accumulation of hydrogen in the bowel; patient should be advised to have a thorough bowel cleansing with a non-fermentable solution.
Other medicines and Gutclear® Syrup
Gutclear® Syrup should not be administered with other laxatives. Caution may be exercised in using Gutclear® Syrup with drugs causing potassium loss like loop diuretics. Gutclear® Syrup can increase digitalis toxicity. Reduction in acidification effect can be observed if broad spectrum antibacterial agents or antacids are administered along with Gutclear® Syrup.
Pregnancy
Gutclear® Syrup should be prescribed only if the potential benefits outweigh the risks to the fetus.
Lactation
No studies are available on the secretion of Gutclear® Syrup in breast milk. There are no data on the presence of lactitol in human or animal milk, the effects on the breastfed infant, or the effects on milk production.
Driving and using machines
Gutclear® Syrup does not affect your ability to drive or use machines.
3. How to Take Gutclear® Syrup
Always take Lactulose exactly as described in this leaflet or as your doctor or pharmacists have told you. Check with your doctor or pharmacist if you are not sure.
Taking this medicine
- Take Gutclear® Syrup from measuring cup.
- You can mix it with fruit juice or water. It is recommended that you drink plenty of fluids (approximately 6-8 glasses throughout the day).
- Swallow the dose immediately. Do not keep it in your mouth as the sugar content may lead to tooth decay, particularly if the syrup is taken for long periods.
- The syrup takes 2 to 3 days to start working.
- After this time, you may be able to reduce the dose you take according to your needs.
Constipation
Adults and adolescents: The starting dose is 30 ml. After this the dose can be adjusted to 15-30ml.
Use in Children: Use of laxatives in children, infants, and babies should be exceptional and under medical supervision because it can influence the normal reflexes for passing stools. Please do not give the syrup to children (under 14 years) before consulting your doctor for prescription and careful supervision.
Gutclear® Syrup is given as a single dose with the morning or evening meal, subsequently adjusted to produce one stool daily.
Hepatic encephalopathy
Adults: The usual starting dose 30-45 ml in 3 to 4 times a day.
Use in Children: No information is available for treatment of children (new-born to 18 years of age) with hepatic encephalopathy.
Use in elderly patients and patients with renal or hepatic insufficiency: No special dosage recommendations exist.
The dose is subsequently adjusted to produce 2 soft stools daily. Doses should be mixed with food or liquid, and it is recommended to drink 1 to 2 glasses of liquid with the meal.
If you take more Gutclear® Syrup than you should
You may develop excessive diarrhoea, which can lead to dehydration. If this occurs, stop taking Gutclear® Syrup and drink plenty of fluids. If you are worried contact your doctor or pharmacist.
If you forget to take Gutclear® Syrup
Take the dose as soon as you remember to take it.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
The most commonly observed adverse effects with Gutclear® Syrup are abdominal cramps, distension or flatulence during the first 10 days of treatment which may likely to disappear after continued administration. The other less frequent side effects in abdominal pain, loose motions, nausea and vomiting, anal itching.
Tell your doctor immediately and stop taking Gutclear® Syrup if you:
Get a serious allergic reaction which causes difficulty in breathing, or swelling of the face, lips, tongue or throat.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top of the home page. By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to Store Gutclear® Syrup
Keep this medicine out of the sight and reach of children.
Do not use Gutclear® Syrup after the expiry date which is stated on the carton. Store below 30°C. Protect from light. Do not freeze.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the Pack and Other Information
What Gutclear® Syrup contains
Each 15 ml contains Lactitol Monohydrate 10 g and Benzoic Acid is 0.0225 g
Gutclear® Syrup is available in 100 or 200ml bottles.
For More Information About This Product
Maxiliv Tablet
1.0 Name of the medicinal product
Glutathione tablet
2.0 Qualitative and quantitative composition
Each film coated tablet contains
Glutathione……………………………………………500 mg
3.0 Dosage form and strength
Tablet 500 mg
4.0 Clinical particulars
4.1 Therapeutic indication
- Alcoholic fatty liver
- Alcoholic liver fibrosis
- Alcoholic liver cirrhosis
- Hepatitis
4.2 Posology and method of administration
Adults: One Maxiliv tablet (500 mg) once or twice daily.
4.3 Contraindications
Patients who show hypersensitivity to reduced glutathione.
4.4 Special warnings and precautions for use
- Administer Maxiliv tablet under the supervision of a doctor.
- Keep out of reach of children.
- If unusual symptoms such as eruption, pale face, blood pressure drop, or difficulty in breathing occur during therapy, discontinue the drug immediately.
4.5 Drugs interactions
Vitamin K, Vitamin B12, Calcium pantothenate and antihistamines can affect the bioavailability of glutathione.
4.6 Use in special populations
Pregnancy and Lactation
Maxiliv tablet should not be administered to a pregnant and lactating woman unless clinical benefit outweighs the risks.
Geriatric
Appropriate dose reduction is required during therapy.
4.7 Effects on ability to drive and use machines
It is not known whether Maxiliv Tablet alters the ability to drive. Do not drive if you experience any symptoms that affect your ability to concentrate and react.
4.8 Undesirable Effects
According to the scientific literature, there are no risks or side effects of oral administration glutathione. However, there have been anecdotal reports of transitory increase in flatulence, gastrointestinal irritation which diminishes after several days of consistent use.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
No specific antidote is available for the treatment of glutathione overdosage. Symptomatic treatment should be provided.
5.0 Pharmacological properties
5.1 Mechanism of Action/ Pharmacodynamic properties
Glutathione is an extremely important cell protectant which directly quenches reactive hydroxyl free radicals, other oxygen-centered free radicals, and radical centers on DNA and other biomolecules. It is an essential cofactor for many enzymes which require thiol-reducing equivalents, and helps to keep redox-sensitive active sites on enzymes in the necessary reduced state. Higher-order thiol cell systems like metallothioneins, thioredoxins, and other redox regulator proteins are ultimately regulated by reduced form of glutathione (GSH) levels and the ratio of GSH/GSSG (oxidized GSH).
GSH/GSSG balance is crucial for homeostasis, stabilizing the cellular biomolecular spectrum, and facilitating cellular performance and survival. GSH and its metabolites also interface with energetics and neurotransmitter syntheses, through several prominent metabolic pathways. GSH availability down-regulates the pro-inflammatory potential of leukotrienes and other eicosanoids. Recently discovered S-nitroso metabolites, generated in-vivo from GSH and NO (nitric oxide) further diversify GSH’s impact on metabolism.
5.2 Pharmacokinetic properties
A 6-month randomized, double-blinded, placebo-controlled trial of oral GSH (250 or 1,000 mg/day) on GSH levels in blood, erythrocytes, plasma, lymphocytes and exfoliated buccal mucosal cells was conducted in 54 non-smoking adults.
GSH levels in blood increased after 1, 3 and 6 months versus baseline at both doses. At 6 months, mean GSH levels increased 30-35 % in erythrocytes, plasma and lymphocytes and 260 % in buccal cells in the high-dose group (P < 0.05). GSH levels increased 17 and 29 % in blood and erythrocytes, respectively, in the low-dose group (P < 0.05). In most cases, the increases were dose and time dependent, and levels returned to baseline after a 1-month washout period. A reduction in oxidative stress in both GSH dose groups was indicated by decreases in the oxidized to reduced glutathione ratio in whole blood after 6 months.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
No known animal toxicology data
7.0 Description
Glutathione (GSH) is a physiological tripeptide- a chain of three amino acid- cysteine, glycine and glutamic acid. It is used for the detoxification of electrophilic metabolites. It is a very efficient free radical scavenger, preventing damage to important cellular components caused by reactive oxygen species such as free radicals and peroxides.
Chemical Name: (2S)-2-Amino-4-[1-(carboxymethyl)carbamoyl-(2R)-2- sulfanylethylcarbamoyl] butanoic acid.
Molecular Formula: C10H17N3O6S
Molecular Weight: 307.32 g/mol
8.0 Pharmaceutical particulars
8.1 List of excipients
8.2 Incompatibilities
8.3 Shelf life
8.4 Special precautions for storage
9.0 Patient counselling information
- Inform your doctor if you have past or current history of asthma.
- Seek immediate medical attention if you have difficulty in breathing after taking the drug.
- Tell your doctor if you are or planning to become pregnant or are breastfeeding.
- Do not take if allergic to glutathione, milk protein or any of its ingredients.
- Do not take if patients had organ transplantation.
- Avoid if suffering from asthma.
- Do not take if patient is Pregnant and breastfeeding women.
12.0 Date of revision
28.11.2024
About leaflet
Please read this leaflet carefully before you start using this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Maxiliv Tablet is and what it is used for
- What you need to know before you take Maxiliv Tablet
- How to take Maxiliv Tablet
- Possible side effects
- How to store Maxiliv Tablet
- Contents of the pack and other information
1. What Maxiliv Tablet is and what it is used for
Maxiliv Tablet contains the active substance glutathione. It is used for the treatment of:
- Alcoholic fatty liver
- Alcoholic liver fibrosis
- Alcoholic liver cirrhosis
- Hepatitis
2. What you need to know before you take Maxiliv Tablet
Do not take Maxiliv Tablet if you:
- Are allergic to glutathione or any of the other ingredients of this medicine.
- Have had an organ transplantation.
- Are pregnant or breastfeeding.
Warnings and precautions:
- Administer Maxiliv Tablet under the supervision of a doctor.
- Keep out of reach of children.
- If you experience unusual symptoms such as rash, pale face, drop in blood pressure, or difficulty in breathing, stop taking the medicine and seek medical attention immediately.
Other medicines and Maxiliv Tablet:
- Vitamin K, Vitamin B12, Calcium pantothenate, and antihistamines can affect the bioavailability of glutathione.
Pregnancy and breastfeeding:
- Maxiliv Tablet should not be used during pregnancy and breastfeeding unless the potential benefit outweighs the risks.
Driving and using machines:
- It is not known whether Maxiliv Tablet affects your ability to drive. Do not drive if you experience symptoms that affect your ability to concentrate and react.
3. How to take Maxiliv Tablet
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
Adults:
Take one Maxiliv Tablet (500 mg) once or twice daily.
If you use more Maxiliv Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Maxiliv Tablet
If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Maxiliv Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
There are no known risks or side effects of oral administration of glutathione. However, there have been anecdotal reports of transient increases in flatulence and gastrointestinal irritation, which diminish after several days of consistent use.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Maxiliv Tablet
- Keep this medicine out of the sight and reach of children.
- Store in a cool, dry place away from direct sunlight.
- Do not use this medicine after the expiry date which is stated on the label and carton after EXP. The expiry date refers to the last day of that month.
- Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Maxiliv Tablet contains:
The active substance is glutathione (500 mg per tablet).
Revised on 11/24
For More Information About This Product
Lornit Syrup
1.0 Generic name
L-Ornithine L-Aspartate syrup
2.0 Qualitative and quantitative composition
Each 5 mL contains:
L-Ornithine-L-Aspartate………………250 mg
Nicotinamide IP ……………………….20 mg
In a flavoured syrup base
Appropriate overages of Vitamins added
3.0 Dosage form and strength
Syrup
4.0 Clinical particulars
4.1 Therapeutic indication
Treatment of associated conditions and sequelae of diseases with impaired hepatic detoxification (e.g. cirrhosis of the liver), when there are symptoms and signs of minimal or overt hepatic encephalopathy.
4.2 Posology and method of administration
Adults: Two teaspoonful twice daily.
Children: One teaspoonful twice daily.
4.3 Contraindications
- Hypersensitivity to LOLA or any other excipients of this product.
- Severe renal insufficiency (serum creatinine value > 3 mg/100 ml).
4.4 Special warnings and precautions for use
- Monitoring of serum and urinary urea levels at regular intervals should be done.
- Should be used during pregnancy only if the potential benefits out-weigh the potential risk to the fetus.
4.5 Drugs interactions
No known drug-drug interaction.
4.6 Use in special populations
Pregnancy & Lactation
The administration in pregnancy and lactation should be avoided. If treatment is nevertheless
thought to be necessary, the benefits and risks should be carefully assessed.
4.7 Effects on ability to drive and use machines
No studies on the effect on the ability to drive and use machines have been performed.
4.8 Undesirable effects
Very rarely side effects like nausea and vomiting occur. These side effects are usually transient and do not necessitate the withdrawal of the drug.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to:medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is no data available for Lornit syrup overdosage. If any patient consumes excess of drug, it should be managed symptomatically.
5.0 Pharmacological properties
5.1 Mechanism of Action/ Pharmacodynamic properties
L-Ornithine-L-Aspartate is a stable salt of the amino acids ornithine and aspartic acid and provides substrates for urea genesis and glutamine synthesis, which are important mechanisms in ammonia detoxification. It is well known that both ornithine and aspartic acid play a key role in the liver metabolism. Ornithine brings ammonia into urea cycle, thereby converting ammonia into urea, a nontoxic substance. The other component of the drug, aspartic acid, not only takes part in an important stage in the reaction sequence involved in the urea cycle, but also features in the tricarboxylic acid cycle as oxaloacetate formed by transamination, thereby improving the energy balance of the diseased liver. Furthermore, aspartic acid promotes natural regeneration of the liver cells by taking part in pryimidine biosynthesis.
Nicotinamide (niacinamide), is the component of two coenzymes nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP) necessary for lipid metabolism, tissue respiration, glycogenolysis, inhibition of very low-density lipoprotein (VLDL) synthesis. It may increase chylomicron triglyceride removal from plasma.
5.3 Pharmacokinetic properties
- L-Ornithine-L-Aspartate is rapidly absorbed and cleaved into L-Ornithine and L-Aspartate.
- Elimination half-life of each amino acid is short, approximately 40 min and bioavailability is 82.2 28% after oral administration.
- Some L-Aspartate appears unchanged in the urine.
Following oral administration nicotinamide is rapidly absorbed (60-76%) with peak plasma time varying from 30-60 min. It mainly metabolizes in the liver with half-life of about 20-45 min. Nicotinamide is excreted in urine (60-88% as unchanged drug).
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
No known animal toxicology data
7.0 Description
Lornit Syrup is a supportive and maintenance therapy during mild to moderate liver-related concerns. It acts as a hepatic protector. It is used as a liver therapy.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on pack
9.0 Patient counselling information
- You will be regularly monitored for blood creatinine and blood/urine urea levels.
- Inform your doctor if you are pregnant, planning a pregnancy, or breastfeeding.
- Do not take this medicine if you are allergic to any of its ingredients.
12.0 Date of revision
23.09.2024
About Leaflet
Please read this leaflet carefully before you start using this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Lornit syrup is and what it is used for
- What you need to know before you take Lornit syrup
- How to take Lornit syrup
- Possible side effects
- How to store Lornit syrup
- Contents of the pack and other information
1. What Lornit Syrup is and what it is used for
Lornit Syrup contains the active substances L-Ornithine L-Aspartate and Nicotinamide. It is used to treat conditions associated with impaired liver detoxification, such as cirrhosis of the liver, and symptoms of hepatic encephalopathy.
2. What you need to know before you take Lornit syrup
Do not take Lornit Syrup if:
- You are allergic to L-Ornithine L-Aspartate, Nicotinamide, or any of the other ingredients of this medicine.
- You have severe renal insufficiency (serum creatinine value > 3 mg/100 ml).
Warnings and precautions:
- Regular monitoring of serum and urinary urea levels is recommended.
- Use during pregnancy should only occur if the potential benefits outweigh the risks.
Other medicines and Lornit Syrup:
- No known drug interactions.
Pregnancy and breastfeeding:
- Avoid use during pregnancy and breastfeeding unless deemed necessary by your doctor.
Driving and using machines:
No studies have been conducted on the effects of this medicine on the ability to drive and use machines.
3. How to take Lornit syrup
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
Adults:
- Two teaspoonfuls twice daily.
Children:
- One teaspoonful twice daily.
Shake the bottle well before use. You can take Lornit syrup with or without food.
If you use more Lornit syrup than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Lornit syrup
If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Lornit syrup
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Rarely, side effects such as nausea and vomiting may occur. These are usually temporary and do not require stopping the medication.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Lornit syrup
- Keep this medicine out of the sight and reach of children.
- Store in a cool, dry place.
- Do not use this medicine after the expiry date which is stated on the bottle and carton after EXP. The expiry date refers to the last day of that month.
- Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Lornit syrup contains:
The active substances are L-Ornithine L-Aspartate (250 mg per 5 mL) and Nicotinamide (20 mg per 5 mL).
Revised on 11/24
For More Information About This Product
Lornit 500 mg Tablet
1.0 Generic name
L-Ornithine L-Aspartate tablet
2.0 Qualitative and quantitative composition
Each film coated tablet contains:
L-Ornithine L-Aspartate 150mg/ 500 mg
Excipients q.s
3.0 Dosage form and strength
Tablet 150 mg/ 500 mg
4.0 Clinical particulars
4.1 Therapeutic indication
Treatment of associated conditions and sequelae of diseases with impaired hepatic detoxification (e.g. cirrhosis of the liver), when there are symptoms and signs of minimal or overt hepatic encephalopathy.
4.2 Posology and method of administration
2-3 tablets 3-4 times per day can be administered in mild to moderate liver disorders.
4.3 Contraindications
- Hypersensitivity to LOLA or any other excipients of this product.
- Severe renal insufficiency (serum creatinine value > 3 mg/100 ml).
4.4 Special warnings and precautions for use
- Monitoring of serum and urinary urea levels at regular intervals should be done.
- Should be used during pregnancy only if the potential benefits out-weigh the potential risk to the fetus.
4.5 Drugs interactions
No known drug-drug interaction.
4.6 Use in special populations
Pregnancy & Lactation
The administration in pregnancy and lactation should be avoided. If treatment is nevertheless
thought to be necessary, the benefits and risks should be carefully assessed.
4.7 Effects on ability to drive and use machines
No studies on the effect on the ability to drive and use machines have been performed
4.8 Undesirable effects
Very rarely side effects like nausea and vomiting occur. These side effects are usually transient and do not necessitate the withdrawal of the drug.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is no data available for LOLA tablet overdosage. If any patient consumes excess of drug, it should be managed symptomatically.
5.0 Pharmacological properties
5.1 Mechanism of Action
L-Ornithine-L-Aspartate is a stable salt of the amino acids ornithine and aspartic acid and provides substrates for urea genesis and glutamine synthesis, which are important mechanisms in ammonia detoxification.
5.2 Pharmacodynamic properties
It is well known that both ornithine and aspartic acid play a key role in liver metabolism. Ornithine is the starting point of ammonia detoxification. It brings ammonia into the urea cycle, in which ammonia is converted into a non-toxic substance urea. It activates ornithine transcarbamoylase and carbamoyl phosphate synthetase and acts as a substrate for urea genesis. Hence, LOLA can activate the periportal urea cycle in the liver. The other component of the drug, aspartic acid, not only represents an important stage in the reaction sequence involved in the urea cycle, but also features in the tricarboxylic acid cycle as oxaloacetate formed by transamination, thereby improving the energy balance of diseased liver. Furthermore, aspartic acid promotes natural regeneration of liver cells by taking part in pyrimidine biosynthesis.
After conversion to α-ketoglutarate, aspartate and ornithine, act as carbon sources for perivenous glutamine synthesis. LOLA up-regulates glutamine synthesis in the skeletal muscle by substrate provision for glutamine synthetase. Ammonia is consumed during urea formation and glutamine synthesis, and thereby LOLA decreases blood ammonia levels.
5.3 Pharmacokinetic properties
- L-Ornithine-L-Aspartate is rapidly absorbed and cleaved into L-Ornithine and L-Aspartate.
- Elimination half-life of each amino acid is short, approximately 40 min and bioavailability is 82.2 28% after oral administration.
- Some L-Aspartate appears unchanged in the urine.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
No known animal toxicology data
7.0 Description
L-Ornithine L-aspartate (LOLA) is a mixture of two endogenous essential amino acids, L-ornithine and L-aspartate. Being a stable salt of L-ornithine and L-aspartate, LOLA readily dissociates into its constituent amino acids that are readily absorbed by active transport, distributed and metabolized.
Molecular formula: C9H19N3O6
Molecular mass: 265.26 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on pack
9.0 Patient Counselling Information
- You will be regularly monitored for blood creatinine and blood/urine urea levels.
- Inform your doctor if you are pregnant, planning a pregnancy, or breastfeeding.
- Do not take this medicine if you are allergic to any of its ingredients.
12.0 Date of revision
23.09.2024
About Leaflet
Please read this leaflet carefully before you start using this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
1. What Lornit is and what it is used for
2. What you need to know before you use Lornit
3. How to use Lornit
4. Possible side effects
5. How to store Lornit
6. Contents of the pack and other information
1. What Lornit is and what it is used for
Lornit contains the active substance L-Ornithine L-Aspartate. It is used to treat conditions associated with impaired liver detoxification, such as cirrhosis of the liver, when there are symptoms and signs of minimal or overt hepatic encephalopathy.
2. What you need to know before you take Lornit
Do not take Lornit if:
- You are allergic to L-Ornithine L-Aspartate or any of the other ingredients of this medicine.
- You have severe renal insufficiency (serum creatinine value > 3 mg/100 ml).
Warnings and precautions:
- Regular monitoring of serum and urinary urea levels is necessary.
- Use during pregnancy should only be if the potential benefits outweigh the potential risks to the fetus.
Pregnancy and breastfeeding:
Driving and using machines:
- Avoid using Lornit during pregnancy and breastfeeding unless deemed necessary by your doctor. Driving and using machines:
No studies have been performed on the effects of Lornit on the ability to drive and use machines.
3. How to take Lornit
Always take this medicine exactly as your doctor has told you. Check with your doctor if you are not sure.
Recommended dose:
- 2-3 tablets, 3-4 times per day for mild to moderate liver disorders.
If you use more Lornit than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Lornit
If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Lornit
Do not stop your treatment even if you feel better unless told to do so by your doctor.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Very rare side effects:
- Nausea and vomiting, which are usually transient and do not require stopping the medication.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com in and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Lornit
- Keep this medicine out of the sight and reach of children.
- Do not store above 25°C. Do not freeze.
- Do not use this medicine after the expiry date which is stated on the label and carton after EXP.
- The expiry date refers to the last day of that month.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Lornit contains:
- The active substance is L-Ornithine L-Aspartate (150 mg or 500 mg per tablet).
For More Information About This Product
Zosa L Capsules
1.0 Name of the medicinal product
Esomeprazole (GR) & Levosulpiride (PR) Capsules
2.0 Qualitative and quantitative composition
Each Hard Gelatin Capsule Contains:
Esomeprazole Magnesium USP (Trihydrate)
equivalent to Esomeprazole ……. ….. 40 mg
(as gastro-resistant pellets)
Levosulpiride ………………………… 75 mg
(as prolonged release pellets)
3.0 Pharmaceutical form and strength
Capsule Esomeprazole (40 mg) & Levosulpiride (75 mg)
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of gastro-esophageal reflux disease (GERD) in patients who do not respond to PPI (proton-pump inhibitor) alone.
4.2 Posology and method of administration
1 capsule to be administered once daily. Zosa L Capsules should be administered on empty stomach, preferably in the morning or at least 1 hour prior to meal. The capsules should be swallowed whole with water and not to be opened, chewed or crushed. Or, as prescribed by the physician
Pediatric Patients
Safety and efficacy of esomeprazole with levosulpiride combination therapy has not been established in paediatric patients. Thus, Zosa L Capsules are not recommended for use in children and adolescents below 18 years of age.
Geriatric Patients
No dosage adjustment is generally necessary in the elderly patients with normal renal function, but dose should be reduced if there is evidence of renal impairment. Elderly patients are more susceptible to postural hypotension, sedation, and extrapyramidal side effects. Thus, caution should be exercised in the elderly population while on Zosa L therapy.
Renal Impairment Patients
Zosa L Capsules should be used with caution and dose/dosage frequency may need to be reduced depending on the severity of the renal dysfunction. Zosa L Capsules are contraindicated in patients with severe renal impairment.
Hepatic Impairment Patients
With esomeprazole, dose adjustment is not required in patients with mild to moderate liver impairment. For patients with severe liver impairment, a maximum dose of 20 mg esomeprazole should not be exceeded. There is no information available on use of levosulpiride in patients with hepatic dysfunction. Thus, as a precautionary measure, Zosa L Capsules should be avoided in patients with hepatic impairment.
4.3 Contraindications
- Patients with known hypersensitivity to esomeprazole or to any substituted benzimidazole derivative or to levosulpiride or to any component of the formulation.
- In patients receiving rilpivirine-containing products.
- Gastrointestinal bleeding and intestinal obstruction.
- Severe renal or hepatic insufficiency.
- Porphyrias.
- Alcohol intoxication.
- Certain tumors like phaeochromocytoma and pituitary prolactinoma.
- Concurrent use with levodopa or other antiparkinson drugs (including ropinirole).
4.4 Special warnings and precautions for use
Esomeprazole
Gastric Malignancy: In the presence of any alarm symptom (e.g., significant unintentional weight loss, recurrent vomiting, dysphagia, haematemesis or melaena) and when gastric ulcer is suspected or present, malignancy should be excluded, as treatment with esomeprazole may alleviate symptoms and delay diagnosis.
Helicobacter Pylori Eradication: When prescribing esomeprazole for eradication of Helicobacter pylori, possible drug interactions for all components in the triple therapy should be considered. Clarithromycin is a potent inhibitor of CYP3A4 and hence contraindications and interactions for clarithromycin should be considered when the triple therapy is used in patients concurrently taking other drugs metabolised via CYP3A4, such as cisapride.
Gastrointestinal Infections/Gastritis: Treatment with proton pump inhibitors (PPIs) may lead to a slightly increased risk of gastrointestinal infections such as Salmonella and Campylobacter. Atrophic gastritis has been noted occasionally in gastric corpus biopsies from patients treated long-term with omeprazole, of which esomeprazole is an enantiomer.
Clostridium Difficile-Associated Diarrhea (CDAD): Published observational studies suggest that PPI therapy like esomeprazole may be associated with an increased risk of Clostridium difficile-associated diarrhea, especially in hospitalized patients. This diagnosis should be considered for diarrhea that does not improve. Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated.
Absorption of Vitamin B12: Esomeprazole, like all acid-blocking medicines, may reduce the absorption of vitamin B12 (cyanocobalamin) due to hypo- or achlorhydria. This should be considered in patients with reduced body stores or risk factors for reduced vitamin B12 absorption on long-term therapy.
Risk of Bone Fracture: Several published observational studies suggest that PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. The risk of fracture was increased in patients who received high-dose (defined as multiple daily doses), and long-term PPI therapy (a year or longer). Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated. Patients at risk for osteoporosis-related fractures should be managed according to established treatment guidelines and they should have an adequate intake of vitamin D and calcium.
Subacute Cutaneous Lupus Erythematosus (SCLE): PPIs have been associated with cases of SCLE, although very infrequently. If lesions occur, especially in sun-exposed areas of the skin, and if accompanied by arthralgia, the patient should seek medical help promptly and esomeprazole therapy should be stopped immediately. The occurrence of SCLE with previous PPI treatment may increase the risk of SCLE with other PPIs.
Hypomagnesemia: Hypomagnesemia (symptomatic/asymptomatic), has been reported rarely in patients treated with PPIs for at least three months, in most cases after a year of therapy. Serious adverse events include tetany, arrhythmias, and seizures. In most patients, treatment of hypomagnesemia required magnesium replacement and discontinuation of the PPI. For patients expected to be on prolonged treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g., diuretics), it is recommended to monitor magnesium levels prior to initiation of PPI treatment, and periodically thereafter.
Levosulpiride
History of Breast Cancer: Levosulpiride may increase prolactin levels. Therefore, caution should be exercised and patients with a history or a family history of breast cancer should be closely monitored during levosulpiride therapy.
Prolongation of the QT Interval: Levosulpiride induces a prolongation of the QT interval. This effect is known to potentiate the risk of serious ventricular arrhythmias such as torsade de pointes. Levosulpiride should be used with caution in patients with cardiovascular disease or with a family history of QT prolongation.
Gastrointestinal Disorders: Levosulpiride should not be used when gastrointestinal stimulation of motility can be harmful e.g., in presence of gastrointestinal hemorrhage, mechanical obstructions or perforations.
Drugs Acting on CNS: Caution is advised when levosulpiride is administered concomitantly with other centrally acting drugs.
Alcohol: Concomitant intake of alcohol should be avoided during levosulpiride therapy as there is an increased chance of sedation.
Smoking: Smoking increases metabolism of the drug and thus, require higher dose of levosulpiride.
Parkinson’s Disease: In patient with Parkinson's disease, levosulpiride use should be avoided and an alternative drug therapy should be considered.
Convulsions: Cases of convulsions, sometimes in patients with no previous history, have been reported. In patients requiring levosulpiride who are receiving anticonvulsant therapy, the dose of the anticonvulsant should not be changed.
Anticholinergic Effects: Levosulpiride has an anticholinergic effect and, therefore, should be used with caution in patients with a history of glaucoma, ileus, congenital digestive stenosis, urine retention or hyperplasia of the prostate.
Hypertensive Patients: Levosulpiride should be used with caution in hypertensive patients, especially in the elderly population, due to the risk of hypertensive crisis. Patients should be adequately monitored.
4.5 Interaction with other medicinal products and other forms of interaction Esomeprazole
1) Interference with Antiretroviral Therapy Reduced Concentrations of Atazanavir and Nelfinavir: Concomitant use of atazanavir and nelfinavir with PPIs is not recommended. Co-administration of atazanavir with PPIs is expected to substantially decrease atazanavir plasma concentrations and may result in a loss of therapeutic effect and the development of drug resistance. If the combination of atazanavir with a PPI is unavoidable, close clinical monitoring is recommended in combination with an increase in the dose of atazanavir to 400 mg with 100 mg of ritonavir; esomeprazole 20 mg should not be exceeded.
Increased Concentrations of Saquinavir: Co-administration of saquinavir with PPIs is expected to increase saquinavir concentrations, which may increase toxicity and require dose reduction. Omeprazole, of which esomeprazole is an enantiomer, has been reported to interact with other antiretroviral drugs, too. The clinical importance and the mechanisms behind these interactions are not always known. Increased gastric pH during omeprazole treatment may change the absorption of the antiretroviral drug. Other possible interaction mechanisms are via CYP 2C19.
2) Drugs for Which Gastric PH Can Affect Bioavailability (ketoconazole, atazanavir, iron salts, erlotinib, mycophenolate mofetil, digoxin) Esomeprazole inhibits gastric acid secretion. Therefore, esomeprazole may interfere with the absorption of drugs where gastric pH is an important determinant of bioavailability. Like with other drugs that decrease intragastric acidity, the absorption of drugs such as ketoconazole, atazanavir, iron salts, erlotinib, and mycophenolate mofetil (MMF) can decrease, while the absorption of drugs such as digoxin can increase during treatment with esomeprazole. Digoxin: Concomitant treatment with omeprazole (20 mg daily), of which esomeprazole is an enantiomer, and digoxin in healthy subjects increased the bioavailability of digoxin by 10%. Co-administration of digoxin with esomeprazole is expected to increase the systemic exposure of digoxin. Therefore, patients may need to be monitored when digoxin is taken concomitantly with esomeprazole.
Mycophenolate Mofetil (MMF): Co-administration of omeprazole in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving esomeprazole and MMF. Use esomeprazole with caution in transplant patients receiving MMF.
3) Effects on Hepatic Metabolism/Cytochrome P-450 Pathways Esomeprazole is extensively metabolized in the liver by CYP 2C19 and CYP 3A4. In-vitro and in-vivo studies have shown that esomeprazole is not likely to inhibit CYPs 1A2, 2A6, 2C9, 2D6, 2E1, and 3A4. No clinically relevant interactions with drugs metabolized by these CYP enzymes would be expected. Drug interaction studies have shown that esomeprazole does not have any clinically significant interactions with quinidine, clarithromycin, or amoxicillin.
Warfarin: Patients treated with PPIs and warfarin concomitantly may need to be monitored for increases in INR (International Normalized Ratio) and prothrombin time. Increases in INR and prothrombin time may lead to abnormal bleeding.
Clopidogrel: Avoid concomitant use of esomeprazole with clopidogrel. Clopidogrel is a prodrug. Inhibition of platelet aggregation by clopidogrel is entirely due to an active metabolite. The metabolism of clopidogrel to its active metabolite can be impaired by use of concomitant medications, such as esomeprazole, that inhibit CYP2C19 activity. Concomitant use of clopidogrel with esomeprazole 40 mg reduces the pharmacological activity of clopidogrel. As a precaution, concomitant use of esomeprazole and clopidogrel should be discouraged or when using esomeprazole consider alternative anti-platelet therapy.
Diazepam: Esomeprazole may potentially interfere with CYP2C19, the major esomeprazole metabolizing enzyme. Co-administration of esomeprazole and diazepam, a CYP2C19 substrate, resulted in a 45% decrease in clearance of diazepam.
Phenytoin: Concomitant administration of esomeprazole 40 mg resulted in a 13% increase in trough plasma levels of phenytoin in epileptic patients. It is recommended to monitor the plasma concentrations of phenytoin when treatment with esomeprazole is introduced or withdrawn.
Cilostazol: Omeprazole as well as esomeprazole act as inhibitors of CYP2C19. Omeprazole, of which esomeprazole is an enantiomer, given in doses of 40 mg to healthy subjects in a cross-over study, increased Cmax and AUC for cilostazol by 18% and 26% respectively, and one of its active metabolites by 29% and 69%, respectively. Cisapride: In healthy volunteers, concomitant administration of esomeprazole 40 mg resulted in a 32% increase in area under the plasma concentration-time curve (AUC) and a 31% prolongation of elimination half-life (t½), but no significant increase in peak plasma levels of cisapride. The slightly prolonged QTc interval observed after administration of cisapride alone, was not further prolonged when cisapride was given in combination with esomeprazole. Voriconazole: Concomitant administration of esomeprazole and a combined inhibitor of CYP 2C19 and CYP3A4, such as voriconazole, may result in a more than doubling of the esomeprazole exposure. Dose adjustment of esomeprazole is not normally required. However, in patients with Zollinger-Ellison's Syndrome, who may require higher doses (up to 240 mg/day), dose adjustment may be considered. Rifampicin: Drugs known to induce CYP2C19 or CYP3A4 or both (such as rifampin) may lead to decreased esomeprazole serum levels. Avoid concomitant use of rifampin with esomeprazole. St. John's Wort: Omeprazole, of which esomeprazole is an enantiomer, has been reported to interact with St. John's wort, an inducer of CYP3A4. Avoid concomitant use of St. John's wort with esomeprazole.
4) Concomitant Administration with Other Drugs
Tacrolimus: Concomitant administration of esomeprazole and tacrolimus may increase the serum levels of tacrolimus.
Combination Therapy with Clarithromycin: Co-administration of esomeprazole, clarithromycin, and amoxicillin has resulted in an increase in plasma levels of esomeprazole and 14-hydroxyclarithromycin.
Methotrexate: Concomitant administration of PPIs and methotrexate (primarily at high dose) may elevate and prolong serum levels of methotrexate and/or its metabolite hydroxymethotrexate, leading to a risk of methotrexate toxicity. In high-dose methotrexate administration, a temporary withdrawal of the PPI may be considered in some patients.
5) Drug / Laboratory Tests Interactions
Interactions with Investigations of Neuroendocrine Tumors: Serum chromogranin A (CgA) levels increase secondary to drug-induced decreases in gastric acidity. The increased CgA level may cause false positive results in diagnostic investigations for neuroendocrine tumors. To avoid this interference, esomeprazole treatment should be stopped for at least 5 days before CgA measurements; consider repeating the test if initial CgA levels are high. Levosulpiride
Antacids and Sucralfate: Bioavailability of levosulpiride is reduced if it is taken concomitantly with sucralfate and aluminum/magnesium-containing antacids. So, these medicines should not be taken along with levosulpiride. There should be a minimum 2 hour time lag between the two medicines. Anticholinergic Drugs, Narcotics and
Analgesic Drugs: The effect of levosulpiride on gastrointestinal motility can be antagonized by these drugs. Antihypertensive Drugs: Concomitant use of levosulpiride may enhance the hypotensive effects of these drugs.
Anticholinergic Drugs: Concomitant administration may cause increase in incidence of anticholinergic side effects.
Levodopa/Antiparkinson Drugs (including ropinirole): There is reciprocal antagonism of effects between levodopa or antiparkinson drugs (including ropinirole) and levosulpiride. Levodopa reduces effects of levosulpiride; conversely, levosulpiride may decrease the efficacy of levodopa in the management of Parkinson’s disease. Thus, concomitant use of these drugs are contraindicated.
Atomoxetine, Antiarrhythmics, Terfenadine, Chloroquine, Quinine, Cisapride, and Drugs Causing Hypokalemia (corticosteroids, laxatives, and diuretics like furosemide): Concurrent use of levosulpiride with these drugs may cause arrhythmia, especially prolonged QT interval.
Alcohol: Levosulpiride can potentiate the cognitive and motor effects of alcohol. Thus, concurrent use should be avoided.
Lithium: Increased risk of extrapyramidal effects. Discontinuation of both drugs is recommended at first signs of neurotoxicity.
4.6 Use in special populations
Pregnancy Zosa L capsules are not recommended for use in pregnant women. Breast-feeding Zosa L capsules should not be used during breast feeding. Accordingly, a decision should be made whether to discontinue nursing or to discontinue/abstain from drug therapy, taking into account the benefit of the drug to the mother.
4.7 Effects on ability to drive and use machines
Patients should avoid driving or operating machinery or not to engage in activities that require full mental alertness.
4.8 Undesirable effects
Esomeprazole
Acute kidney injury as an adverse drug reaction reported with the use of proton pump inhibitors.
The most frequently reported (≥ 1%) adverse reactions with esomeprazole were headache, diarrhea, nausea, flatulence, abdominal pain, constipation, and dry mouth. Additional adverse reactions that were reported as possibly or probably related to esomeprazole with an incidence < 1% are as follows:
Body as a Whole: Enlarged abdomen, allergic reaction, asthenia, back pain, chest pain, substernal chest pain, facial edema, peripheral edema, hot flushes, fatigue, fever, flu-like symptoms, generalized edema, leg edema, malaise, pain, rigors.
Cardiovascular: Flushing, hypertension, tachycardia.
Endocrine: Goiter.
Gastrointestinal (GI): Bowel irregularity, constipation aggravated, dyspepsia, dysphagia, GI dysplasia, epigastric pain, eructation, esophageal disorders, frequent stools, gastroenteritis, GI hemorrhage, GI symptoms not otherwise specified, hiccup, melena, mouth disorders, pharyngeal disorders, rectal disorders, increase in serum gastrin, tongue disorders, tongue edema, ulcerative stomatitis, vomiting.
Hearing: Earache, tinnitus.
Hematologic: Anemia, cervical lymphadenopathy, epistaxis, leukocytosis, leukopenia, thrombocytopenia.
Hepatic: Abnormalities in hepatic function, including bilirubinemia, increase in AST (aspartate aminotransferase) and ALT (alanine aminotransferase).
Metabolic/Nutritional: Glycosuria, hyperuricemia, hyponatremia, increased alkaline phosphatase, thirst, vitamin B12 deficiency, weight increase/decrease.
Musculoskeletal: Arthropathy, cramps, fibromyalgia syndrome, hernia, polymyalgia rheumatica.
Nervous System/Psychiatric: Anorexia, apathy, increased appetite, confusion, depression aggravated, dizziness, hypertonia, nervousness, hypoesthesia, impotence, insomnia, migraine, paresthesia, sleep disorder, somnolence, tremor, vertigo, visual field defect.
Reproductive: Dysmenorrhea, menstrual disorders, vaginitis.
Respiratory: Aggravated asthma, coughing, dyspnea, laryngeal edema, pharyngitis, rhinitis, sinusitis.
Skin and Appendages: Acne, angioedema, dermatitis, pruritus, rash, urticaria, sweating.
Special Senses: Otitis media, parosmia, taste loss, taste perversion.
Urogenital: Albuminuria, cystitis, dysuria, fungal infection, hematuria, frequent micturition, moniliasis, polyuria.
Visual: Conjunctivitis, abnormal vision.
Laboratory Abnormalities
The following clinically significant laboratory changes in clinical trials, irrespective of relationship to esomeprazole, were reported in ≤ 1% of patients: Increased creatinine, uric acid, total bilirubin, alkaline phosphatase, ALT, AST, hemoglobin, white blood cell count, platelets, serum gastrin, potassium, sodium, thyroxine and thyroid stimulating hormone. Decreased levels of hemoglobin, white blood cell count, platelets, potassium, sodium, and thyroxine.
Post-Marketing Experience The following adverse reactions have been identified during post-marketing use of esomeprazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic: Agranulocytosis, pancytopenia.
Eye: Blurred vision.
Gastrointestinal: Pancreatitis, stomatitis, microscopic colitis.
Hepatobiliary: Hepatitis with or without jaundice, hepatic failure.
Immune System: Anaphylactic reaction/shock.
Infections and Infestations: GI candidiasis, Clostridium difficile-associated diarrhea.
Metabolism and Nutritional Disorders: Hypomagnesemia.
Musculoskeletal and Connective Tissue: Muscular weakness, myalgia, fractures.
Nervous System: Hepatic encephalopathy, taste disturbance.
Psychiatric: Aggression, agitation, depression, hallucination.
Renal and Urinary: Interstitial nephritis.
Reproductive System and Breast: Gynecomastia.
Respiratory, Thoracic, and Mediastinal: Bronchospasm.
Skin and Subcutaneous Tissue: Alopecia, erythema multiforme, hyperhidrosis, photosensitivity, Stevens-Johnson syndrome, toxic epidermal necrolysis (sometime fatal), cutaneous lupus erythematosus.
Levosulpiride
The following side effects may occur with the use of levosulpiride: Acute muscular dystonia characterized by abnormal movements (twitching, tremor, etc.) of the hands, leg, tongue and facial muscles.
Sedation or drowsiness (because of decrease in sensory inputs to reticular activating system).
Increase in plasma prolactin levels manifested by breast enlargement (gynecomastia), production of milk (galactorrhea) and stopping of menstrual periods (amenorrhea).
Neuroleptic malignant syndrome (characterized by hyperpyrexia, muscle rigidity, increased myoglobin and creatine kinase).
Akathisia (uncontrollable desire to move about without any anxiety).
Tardive dyskinesia, it occurs late in the therapy and its features include involuntary rhythmical movements of face, mouth and jaw. The reason for tardive dyskinesia is synthesis of newer dopamine receptors which are supersensitive to even a small amount of dopamine. This causes a decrease in cholinergic activity in the striatum followed by decrease in gamma-amino butyric acid (GABA) release. This decreased in inhibitory GABA is responsible for increased involuntary motor activity.
Postural hypotension (because of autonomic blockade), tolerance develops to this effect after some time.
Weight gain. Elevated liver transaminases.
Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions via
email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Experience with overdose is limited. There is no specific antidote. Treatment is only symptomatic. Appropriate supportive measures should therefore be instituted, close supervision of vital functions and cardiac monitoring (risk of QT interval prolongation and subsequent ventricular arrhythmias) is recommended until the patient recovers. If severe extrapyramidal symptoms occur anticholinergic drugs should be administrated. Overdose may be treated with alkaline osmotic diuresis and, if necessary, anti-parkinson drugs. Coma needs appropriate nursing, and cardiac monitoring is recommended until the patient recovers.
5.0 Pharmacological properties
5.1 Mechanism of action
Esomeprazole
Esomeprazole is a proton pump inhibitor (PPI) that suppresses gastric acid (hydrochloric acid - HCl) secretion by specific inhibition of the H+/K+-ATPase enzyme system at the secretory surface of the gastric parietal cell. Esomeprazole is a weak base and is concentrated and converted to the active form (i.e., the achiral sulphenamide) in the highly acidic environment of the secretory canaliculi of the parietal cell, where it inhibits the enzyme H+K+-ATPase (the acid/proton pump), and inhibits both basal and stimulated acid secretion. By acting specifically on the proton pump, esomeprazole blocks the final step in acid production, thereby reducing gastric acidity.
Levosulpiride
Prokinetic Effect: Levosulpiride is principally a dopamine D2 antagonist. Dopamine has a direct relaxant effect on the gut by activating muscular D2 receptors. Levosulpiride stimulates gut motility by blocking D2 receptors in lower esophageal sphincter (LES) and stomach. Levosulpiride also acts as an agonist on serotonin 5-HT4 receptors and thus, increases acetylcholine level which leads to increase in GI motility. Antiemetic Effect: Levosulpiride exerts a selective antagonist activity on the D2 receptors on neurons in the CNS (postrema area of 4th ventricle) and thus, produces antiemetic effect. Levosulpiride is also a weak inhibitor of 5-HT3 receptors. Levosulpiride also acts as a moderate agonist at the serotonergic (5-HT4) receptor. This enhances its therapeutic efficacy in gastrointestinal disorders (reduction of nausea and vomiting).
5.2 Pharmacodynamic properties
Esomeprazole
Esomeprazole is the S-isomer of omeprazole. Esomeprazole reduces gastric acid secretion through a specific targeted mechanism of action i.e., inhibition of the acid pump in the gastric parietal cell. Both the R- and S-isomer of omeprazole have similar pharmacodynamic activity.
After oral dosing with esomeprazole 20 mg and 40 mg the onset of effect occurs within one hour. After repeated administration with 20 mg esomeprazole once daily for 5 days, mean peak acid output decreases by 90% when measured 6 to 7 hours after dosing on day five.
After 5 days of oral dosing with 20 mg and 40 mg of esomeprazole, intragastric pH above 4 was maintained for a mean time of 13 hours and 17 hours, respectively over 24 hours in symptomatic GERD patients. The proportion of patients maintaining an intragastric pH above 4 for at least 8, 12 and 16 hours respectively were 76%, 54% and 24% for esomeprazole 20 mg. Corresponding proportions for esomeprazole 40 mg were 97%, 92% and 56%.
Esomeprazole increases the mean fasting gastrin level in a dose-related manner. This increase reached a plateau within two to three months of therapy and returned to baseline levels within four weeks after discontinuation of therapy.
Levosulpiride
Levosulpiride, levo-enantiomer (biologically active form) of sulpiride, is a benzamide derivative. The levo-enantiomer shows better/similar pharmacological actions and lower incidence of toxic effects than both dextro as well as the racemic forms of the drug. Due to its peripheral anti-dopaminergic action, levosulpiride exhibits gastrokinetic/prokinetic, antiemetic, and anti-dyspeptic effects. Levosulpiride act as a modulator of the motor activity of the upper digestive tract. Levosulpiride accelerates gastric emptying and improves gastrointestinal (GI) symptoms such as heart burn, regurgitation, etc. by selectively inhibitingdopaminergic receptors (D2) in the submucosal and myoenteric plexus of the gastrointestinal tract (GIT) and chemoreceptor trigger zone (CTZ) of the central nervous system (CNS).
5.3 Pharmacokinetic Properties
Esomeprazole
Absorption: Zosa L Capsules contains esomeprazole as a gastro-resistant tablet. This is necessary because, like other PPIs, esomeprazole is acid-labile drug. Absorption of esomeprazole therefore, begins only after the tablet leaves the stomach. Esomeprazole is rapidly absorbed after oral administration. Onset of effect occurs within one hour after oral dosing with esomeprazole 20 mg and 40 mg. Peak plasma levels (Cmax) occur approximately 1 to 2 hours (Tmax) after oral dose. The Cmax increases proportionally when the dose is increased, and there is a three-fold increase in the area under the plasma concentration-time curve (AUC) from 20 to 40 mg. At repeated once daily dosing with 40 mg, the systemic bioavailability is approximately 90% compared to 64% after a single dose of 40 mg. Effect of Food: Food intake both delays and decreases the absorption of esomeprazole. Thus, esomeprazole should be administered at least one hour before meal. Distribution: Esomeprazole is 97% bound to plasma proteins. Plasma protein binding is constant over the concentration range of 2 to 20 μmol/l. The apparent volume of distribution at steady state in healthy volunteers is approximately 16 liters. Metabolism: Esomeprazole is extensively metabolized in the liver by the cytochrome P450 (CYP) enzyme system. The metabolites of esomeprazole lack antisecretory activity. The major part of esomeprazole's metabolism is dependent upon the CYP2C19 isoenzyme, which forms the hydroxy and desmethyl metabolites. The remaining amount is dependent on CYP3A4 which forms the sulphone metabolite. Excretion: Total plasma clearance is about 17 l/h after a single dose and about 9 l/h after repeated administration. The plasma elimination half-life is about 1.3 hours after repeated once daily dosing. Esomeprazole is completely eliminated from plasma with no tendency for accumulation during once-daily administration. Approximately 80% of an oral dose of esomeprazole is excreted as inactive metabolites in the urine, and the remainder is found as inactive metabolites in the feces. Less than 1% of parent drug is excreted in the urine.
Levosulpiride
Pharmacokinetics of levosulpiride in sustained release formulation is not available. Conventional formulation of levosulpiride (i.e., immediate release) has following pharmacokinetic properties: Levosulpiride when administered orally exhibited linear pharmacokinetic properties over the dose range of 25 to 100 mg. The bioavailability of levosulpiride when given orally is about 30%. Sulpiride is slowly absorbed from the gastrointestinal tract with peak plasma concentrations are attained 3 to 6 hours after oral dose. After repeated administration, steady state was reached on day 4 of multiple dosing. Sulpiride is about 40% bound to plasma proteins and is reported to have a plasma half-life of about 8 to 9 hours. Levosulpiride is mainly excreted through the renal route.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Esomeprazole
Carcinogenicity: The carcinogenic potential of esomeprazole magnesium was assessed using studies of omeprazole, of which esomeprazole is an enantiomer. In two 24-month oral carcinogenicity studies in rats, omeprazole at daily doses of 1.7 mg/kg/day, 3.4 mg/kg/day, 13.8 mg/kg/day, 44 mg/kg/day, and 140.8 mg/kg/day (about 0.4 to 34 times the human dose of 40 mg/day expressed on a body surface area basis) produced gastric ECL cell carcinoids in a dose-related manner in both male and female rats; the incidence of this effect was markedly higher in female rats, which had higher blood levels of omeprazole. In addition, ECL cell hyperplasia was present in all treated groups of both sexes. In one of these studies, female rats were treated with 13.8 mg omeprazole/kg/day (about 3.4 times the human dose of 40 mg/day on a body surface area basis) for 1 year, then followed for an additional year without the drug. No carcinoids were seen in these rats. An increased incidence of treatment-related ECL cell hyperplasia was observed at the end of 1 year (94% treated vs. 10% controls). By the second year the difference between treated and control rats was much smaller (46% vs. 26%) but still showed more hyperplasia in the treated group. Mutagenesis: Esomeprazole was negative in the Ames mutation test, in the in vivo rat bone marrow cell chromosome aberration test, and the in vivo mouse micronucleus test. Esomeprazole, however, was positive in the in vitro human lymphocyte chromosome aberration test. Impairment of Fertility: The potential effects of esomeprazole on fertility and reproductive performance were assessed using omeprazole studies. Omeprazole at oral doses up to 138 mg/kg/day in rats (about 34 times the human dose of 40 mg/day on a body surface area basis) was found to have no effect on reproductive performance of parental animals. Reproduction Studies: Reproduction studies have been performed in rats at oral doses up to 280 mg/kg/day (about 68 times an oral human dose of 40 mg on a body surface area basis) and in rabbits at oral doses up to 86 mg/kg/day (about 42 times an oral human dose of 40 mg on a body surface area basis) and have revealed no evidence of impaired fertility or harm to the fetus due to esomeprazole.
Levosulpiride
The values expressed as LD50 acute toxicity after oral administration in mice, rats and rabbits were 2450 mg / kg, 2600 mg / kg and greater than 1500 mg / kg. Subacute toxicity tests were conducted by administering the active ingredient in rat, rabbit and dog, daily, for 12-13 weeks. The appearance of any toxic symptoms was not observed at doses of: 25 mg / kg s.c. and 300 mg / kg p.o. in the rat; 250 mg / kg p.o. and 12.5 mg / kg i.m. in rabbits; and 50 and 100 mg / kg p.o. in the dog. To evidentiate the chronic toxicity after administration of the drug for 180-190 days, the following doses were well tolerated: 100 mg / kg p.o. and 20 mg / kg s.c. in the rat; 10 mg / kg i.m. in rabbits; and 20 mg / kg p.o. in the dog. Studies performed in rats and mice, administering the medicine at a dose higher than that expected for man, have shown that levosulpiride do not possess carcinogenic properties. Studies carried out in rats and rabbits have shown that the medicine is not teratogenic. In vitro tests have ruled out that levosulpiride possesses mutagenic properties.
7.0 Description
Each capsule of Zosa L contains 40 mg of esomeprazole (in a gastro-resistant form) and 75 mg of levosulpiride (in a prolonged release form) for oral administration in adults.
Esomeprazole Magnesium
Esomeprazole magnesium is the magnesium salt of esomeprazole, the S-isomer of omeprazole, with gastric proton pump inhibitor activity. Esomeprazole magnesium salt is off-white to pale cream colored powder. It is slightly soluble in water.
Molecular Weight: 713.12 g/mol.
Molecular Formula: C34H36MgN6O6S2.
Chemical Name: 5-methoxy-2-[(S)-[(4-methoxy-3,5-dimethyl-2-pyridyl)methyl] sulfinyl]benzimidazole, magnesium salt (2:1).
Levosulpiride
Levosulpiride is the active levorotatory enantiomer of the racemic drug sulpiride, a substituted benzamide. Levosulpiride is a dopaminergic antagonist with prokinetic, antiemetic, antidepressant, and antipsychotic properties. Molecular Weight: 341.4 g/mol.
Molecular Formula: C15H23N3O4S.
Chemical Name: N-[[(2S)-1-ethylpyrrolidin-2-yl]methyl]-2-methoxy-5-
sulfamoylbenzamide.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf Life
Refer on pack
8.3 Packaging Information
1 Blister strip of 15 capsules
8.4 Storage and handing Instructions
Store protected from light & moisture at a temperature not exceeding 30°.
Keep out of reach of children.
9.0 Patient counselling information
- Instruct patients to take Zosa L Capsules exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting to your doctor.
- Instruct patients to take Zosa L capsules on empty stomach, preferably in the morning or at least 1 hour prior to meal. The capsules should be swallowed whole with water and not to be opened, chewed or crushed.
- If you miss a dose, take it as soon as possible. If it is almost time for your next dose, do not take the missed dose. Take the next dose at your regular time. Do not take 2 doses/capsules at the same time to make up for the missed dose.
- Instruct patients not to use this medicine during pregnancy.
- Advise nursing mothers to avoid use of this medicine during lactation or not to breastfeed their infants while on drug therapy.
- This medicine is not recommended for use in children.
- Instruct patients not to share this medication with other people even though symptoms are similar. It may harm them.
- Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Zosa L Capsules and certain other medicines can interact with each other causing serious side effects.
12.0 Date of Revision
28.08.2024
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Zosa-L is and what it is used for
- What you need to know before you take Zosa-L
- How to take Zosa-L
- Possible side effects
- How to store Zosa-L
- Contents of the pack and other information
1. What Zosa-L is and what it is used for
The active ingredients of Zosa-L capsules are Esomeprazole and Levosulpiride.
Esomeprazole belongs to a group of medicines called Proton Pump Inhibitors (PPIs). It acts by reducing the amount of acid made by the stomach.
Levosulpiride belongs to the class of antipsychotic medicines. However, it is primarily used for its gut motility properties i.e., to improve food movement and treats disorders of the stomach and intestines. This medicine works by inhibiting the action of certain specific neurotransmitters in the brain and specific regions of the stomach and the intestine.
Zosa-L capsules are used to treat:
- Gastro-esophageal reflux disease (GERD). GERD is commonly referred to as inflammation of the gullet caused by acid and associated with heartburn. Heartburn is a burning feeling rising from the stomach or lower chest up towards the neck.
2. What you need to know before you take Zosa-L
Do not take Zosa-L if you:
- Patients with known hypersensitivity to esomeprazole or to any substituted benzimidazole derivative or to levosulpiride or to any component of the formulation.
- have Prolactin-releasing pituitary tumour (prolactinoma).
- have hepatic and renal impairment, epilepsy, manic states, hyperprolactinaemia, porphyria, mammary dysplasia, malignant mastopathies, pheochromocytoma.
- have known existing prolongation of cardiac conduction intervals, particularly QTc, patients with significant electrolyte disturbances or underlying cardiac diseases such as congestive heart failure.
- gastro-intestinal haemorrhage, mechanical obstruction or perforation.
Warnings and precautions
Talk to your doctor or pharmacist before taking Zosa-L if you:
- have gastric malignancy such as ulcers, gastrointestinal infections, or gastritis
- have Clostridium Difficile-Associated Diarrhea (CDAD)
- are at risk for osteoporosis-related fractures
- are due to have a specific blood test (Chromogranin A).
Zosa-L may hide the symptoms of other diseases. Therefore, if any of the following happen to you before you start taking Zosa-L or while you are taking it, talk to your doctor straight away:
- You lose a lot of weight for no reason and have problems swallowing.
- You get stomach pain or indigestion.
- You begin to vomit food or blood.
- You pass black stools (blood-stained faeces).
If you have been prescribed Zosa-L “on demand” you should contact your doctor if your symptoms continue or change in character.
Taking a proton pump inhibitor like Zosa-L, especially over a period of more than one year, may slightly increase your risk of fracture in the hip, wrist or spine. Tell your doctor if you have osteoporosis or if you are taking corticosteroids (which can increase the risk of osteoporosis).
Rash and skin symptoms
If you get a rash on your skin, especially in areas exposed to the sun tell your doctor as soon as you can, as you may need to stop your treatment with Zosa-L. Remember to also mention any other ill-effects like pain in your joints.
Serious skin rashes have occurred in patients taking Zosa-L. The rash can involve ulcers of the mouth, throat, nose, genitals and conjunctivitis (red and swollen eyes). These serious skin rashes often come after flu-like symptoms such as fever, headache, body ache. The rash may cover large parts of the body with blistering and peeling of the skin. If at any time during the treatment (even after several weeks) you develop a rash or any of these skin symptoms, stop taking this medicine and contact your doctor immediately.
Children under the age of 12 years
Zosa-L is not recommended for children less than 12 years old.
Other medicines and Zosa-L
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
This is because Zosa-L can affect the way some medicines
work and some medicines can have an effect on Zosa-L. Do not take Zosa-L if you are taking a medicine containing Nelfinavir (used to treat HIV infection). Tell your doctor or pharmacist if you are taking any of the following medicines:
- Atazanavir (used to treat HIV infection).
- Saquinavir
- lopidogrel (used to prevent blood clots).
- Ketoconazole, itraconazole or voriconazole (used to treat infections caused by a fungus).
- Erlotinib (used to treat cancer).
- Citalopram, imipramine or clomipramine (used to treat depression).
- Diazepam (used to treat anxiety, relax muscles or in epilepsy).
- Phenytoin (used in epilepsy).
- Medicines that are used to thin your blood, such as warfarin.
- Cilostazol (used to treat intermittent claudication – a pain in your legs when you walk which is caused by an insufficient blood supply).
- Cisapride (used for indigestion and heartburn).
- Digoxin (used for heart problems).
- Methotrexate (a chemotherapy medicine used in high doses to treat cancer)
- Tacrolimus (organ transplantation).
- Rifampicin (used for treatment of tuberculosis).
- St. John’s wort (Hypericum perforatum) (used to treat depression).
If your doctor has prescribed the antibiotics amoxicillin and clarithromycin as well as Zosa-L to treat ulcers caused by Helicobacter pylori infection, it is very important that you tell your doctor about any other medicines you are taking.
Zosa-L with food and drink
You can take the capsules with food or on an empty stomach.
Pregnancy, breast-feeding and fertility If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Your doctor will decide whether you can take Zosa-L during this time.
Levosulpiride may be distributed into breast milk. Therefore, you should not take Zosa-L if you are breast-feeding.
Driving and using machines Zosa-L may have an influence on the ability to drive and use machines.
under treatment may experience numbness, dizziness or involuntary movements (dyskinesia); therefore, they should be advised to avoid driving or operating machines
3. How to take Zosa-L
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
- If you are taking this medicine for a long time, your doctor will want to monitor you (particularly if you are taking it for more than a year).
- If your doctor has told you to take this medicine as and when you need it, tell your doctor if your symptoms change.
How much to take
- Your doctor will tell you how many tablets to take and how long to take them for. This will depend on your condition, how old you are and how well your liver works.
The recommended dose is: Once capsule once daily in adults
Use in children
Zosa-L capsules are not recommended for children below 12 years of age.
Elderly
Dose adjustment is not required in the elderly.
If you take more Zosa-L than you should
If you take more Zosa-L than prescribed by your doctor, talk to your doctor or pharmacist straight away.
If you forget to take Zosa-L
- If you forget to take a dose, take it as soon as you remember it. However, if it is almost time for your next dose, skip the missed dose.
- Do not take a double dose (two doses at the same time) to make up for a forgotten dose.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
If you notice any of the following serious side effects, stop taking Zosa-L and contact a doctor immediately:
- Adverse drug reaction reports show that Proton Pump Inhibitors like Pantoprazole, Omeprazole, Lansoprazole, Esomeprazole, Rabeprazole etc are associated with acute kidney injury
- The most common adverse events were drowsiness/sedation and endocrine effects.
Other side effects include:
Common (may affect up to 1 in 10 people)
- Headache.
- Effects on your stomach or gut: diarrhoea, stomach pain, constipation, wind (flatulence).
- Feeling sick (nausea) or being sick (vomiting).
- Benign polyps in the stomach.
Uncommon (may affect up to 1 in 100 people)
- Swelling of the feet and ankles.
- Disturbed sleep (insomnia).
- Dizziness, tingling feelings such as “pins and needles”, feeling sleepy.
- Spinning feeling (vertigo).
- Dry mouth.
- Changes in blood tests that check how the liver is working.
- Skin rash, lumpy rash (hives) and itchy skin.
- Fracture of the hip, wrist or spine (if Esomeprazole is used in high doses and over long duration).
Rare (may affect up to 1 in 1,000 people)
- Blood problems such as a reduced number of white cells or platelets. This can cause weakness, bruising or make infections more likely.
- Allergic reactions e.g. fever, angioedema and shock
- Low levels of sodium in the blood. This may cause weakness, being sick (vomiting) and cramps.
- Feeling agitated, confused or depressed.
- Taste changes.
- Eyesight problems such as blurred vision.
- Suddenly feeling wheezy or short of breath (bronchospasm).
- An inflammation of the inside of the mouth.
- An infection called “thrush” which can affect the gut and is caused by a fungus.
- Liver problems, including jaundice which can cause yellow skin, dark urine, and tiredness.
- Hair loss (alopecia).
- Skin rash on exposure to sunshine.
- Joint pains (arthralgia) or muscle pains (myalgia).
- Generally feeling unwell and lacking energy.
- Increased sweating.
Very rare (may affect up to 1 in 10,000 people)
- Changes in blood count including agranulocytosis (lack of white blood cells).
- Aggression.
- Seeing, feeling or hearing things that are not there (hallucinations).
- Severe liver problems leading to liver failure and inflammation of the brain.
- Sudden onset of a severe rash or blistering or peeling skin. This may be associated with a high fever and joint pains (Erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms).
- Muscle weakness.
- Severe kidney problems.
- Enlarged breasts in men.
Not known (frequency cannot be estimated from the available data)
- If you are on Zosa-L for more than three months it is possible that the levels of magnesium in your blood may fall. Low levels of magnesium can be seen as fatigue, involuntary muscle contractions, disorientation, convulsions, dizziness or increased heart rate. If you get any of these symptoms, please tell your doctor promptly. Low levels of magnesium can also lead to a reduction in potassium or calcium levels in the blood. Your doctor may decide to perform regular blood tests to monitor your levels of magnesium.
- Inflammation in the gut (leading to diarrhoea).
- Rash, possibly with pain in the joints.
Levosulpiride
Major & minor side effects of Levosulpiride
- Drowsiness
- Breast tenderness
- Irregular menstrual periods
- Gynecomastia
- Constipation
- Abdominal pain and cramps
- Weight gain
- Sleeplessness
- Unusual tiredness and weakness
- Increased salivation
- Decrease in libido
- Fever
- Excessive sweating Change in heart rate
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Zosa-L
- Keep out of reach of children.
- Do not use this medicine after the expiry date which is stated on the blister and the carton after [Exp]. The expiry date refers to the last day of that month.
- Store protected from light & moisture at a temperature not exceeding 30°C.
- Do not throw away medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
Each hard gelatin capsule contains:
Esomeprazole Magnesium Trihydrate IP
equivalent to Esomeprazole…………………………………. 40 mg
(As Gastro-resistant tablet)
Colour: Ferric Oxide (Red) USP-NF
Levosulpiride ……………………………………………..75 mg
(As Prolonged-release uncoated tablet)
Approved colours used in hard gelatin capsule shells.
Presentation/pack size
10 Blister strips of 15 capsules each
For More Information About This Product
Zosa Fast Tablets
1.0 Generic name
Esomeprazole Tablets 40 mg (with Sodium Bicarbonate as buffer)
2.0 Qualitative and quantitative composition
Each fllm coated tablet contains :
Esomeprazole Magnesium Trihydrate IP
equivalent to Esomeprazole 40 mg
Excipients q.s.
Colour : Titanium Dioxide IP
3.0 Dosage form and strength
Tablet 40 mg
4.0 Clinical particulars
4.1 Therapeutic indication
Gastroesophageal Reflux Disease (GERD), erosive reux esophagitis, prevention of relapse of esophagitis and helps in eradication of H.Pylori associated peptic ulcer. It is also indicated for pathological hypersecretory conditions including Zollinger-Ellison syndrome ulcer, risk reduction of NSAID-associated gastric ulcer, and prolonged treatment after I.V. induced prevention of rebleeding of peptic ulcer.
4.2 Posology and method of administration
Adults
Gastroesophageal reflux disease (GERD)
Treatment of erosive reflux esophagitis
40 mg once daily for 4 weeks.
An additional 4 weeks treatment is recommended for patients in whom esophagitis has not healed or who have persistent symptoms.
Long-term management of patients with healed esophagitis to prevent relapse
20 mg once daily
Symptomatic treatment of gastro-oesophageal reux disease (GERD)
20 mg once daily in patients without esophagitis. If symptom control has not been achieved after 4 weeks, the patient should be further investigated. Once symptoms have resolved, subsequent symptom control can be achieved using 20 mg once daily. An on demand regimen taking 20 mg once daily, when needed, can be used. In NSAID treated patients at risk of developing gastric and duodenal ulcers, subsequent symptom control using an on demand regimen is not recommended.
In combination with appropriate antibacterial therapeutic regimens for the eradication of Helicobacter pylori
Healing of Helicobacter pylori associated duodenal ulcer. Prevention of relapse of peptic ulcers in patients with Helicobacter pylori associated ulcers. 20 mg Esomeprazole tablet with 1 g amoxicillin and 500 mg clarithromycin, all twice daily for 7 days.
Patients requiring continued NSAID therapy
Healing of gastric ulcers associated with NSAID therapy : The usual dose is 20 mg once daily. The treatment duration is 4 - 8 weeks. Prevention of gastric and duodenal ulcers associated with NSAID therapy in patients at risk : 20 mg once daily.
Prolonged treatment after I.V. induced prevention of rebleeding of peptic ulcers
40 mg once daily for 4 weeks after I.V. induced prevention of rebleeding of peptic ulcers.
Treatment of Zollinger Ellison syndrome
The recommended initial dosage is Esomeprazole 40 mg twice daily. The dosage should then be individually adjusted and treatment continued as long as clinically indicated. Based on the clinical data available, the majority of patients can be controlled on doses between 80 to 160 mg Esomeprazole daily. With doses above 80 mg daily, the dose should be divided and given twice daily.
Special populations
Renal impairment
Dose adjustment is not required in patients with impaired renal function. Due to limited experience in patients with severe renal insufficiency, such patients should be treated with caution.
Hepatic impairment
Dose adjustment is not required in patients with mild to moderate liver impairment. For patients with severe liver impairment, a maximum dose of 20 mg Esomeprazole tablet should not be exceeded.
Elderly
Dose adjustment is not required in the elderly.
Paediatric population
Adolescents from the age of 12 years
Gastroesophageal reflux disease (GERD)
Treatment of erosive reflux esophagitis
40 mg once daily for 4 weeks.
An additional 4 weeks treatment is recommended for patients in whom esophagitis has not healed or who have persistent symptoms.
Long-term management of patients with healed esophagitis to prevent relapse
20 mg once daily.
Symptomatic treatment of gastroesophageal reflux disease (GERD)
20 mg once daily in patients without esophagitis. If symptom control has not been achieved after 4 weeks, the patient should be further investigated. Once symptoms have resolved, subsequent symptom control can be achieved using 20 mg once daily
Treatment of duodenal ulcer caused by Helicobacter pylori
When selecting appropriate combination therapy, consideration should be given to ofcial national, regional and local guidance regarding bacterial resistance, duration of treatment (most commonly 7 days but sometimes up to 14 days), and appropriate use of antibacterial agents. The treatment should be supervised by a specialist.
The posology recommendation is :
| Weight | Posology |
| 30 - 40 kg | Combination with two antibiotics : Esomeprazole Tablet 20 mg, Amoxicillin 750 mg and Clarithromycin 7.5 mg/kg body weight are all administered together twice daily for one week. |
| > 40 kg | Combination with two antibiotics : Esomeprazole Tablet 20 mg, Amoxicillin 1 g and Clarithromycin 500 mg are all administered together twice daily for one week. |
Children below the age of 12 years
For posology in patients aged 1 to 11 other pharmaceutical forms e.g. sachet should be used.
Method of administration
The tablets should be swallowed whole with liquid. The tablets should not be chewed or crushed. For patients who have difficulty in swallowing the tablets can also be dispersed in half a glass of non-carbonated water. No other liquids should be used as the enteric coating may be dissolved. Stir until the tablets disintegrate and drink the liquid with the pellets immediately or within 30 minutes. Rinse the glass with half a glass of water and drink. The pellets must not be chewed or crushed.
For patients who cannot swallow, the tablets can be dispersed in non-carbonated water and administered through a gastric tube.
4.3 Contraindications
Zosa FAST must not be used in the following conditions :
Hypersensitivity to the active substance, to substituted Benzimidazole or to any of the excipients.
Esomeprazole should not be used concomitantly with Nelfinavir.
4.4 Special warnings and precautions for use
In the presence of any alarm symptom (e.g. signicant unintentional weight loss, recurrent vomiting, dysphagia, haematemesis or melaena) and when gastric ulcer is suspected or present, malignancy should be excluded, as treatment with Esomeprazole may alleviate symptoms and delay diagnosis.
Long term use
Patients on long-term treatment (particularly those treated for more than a year) should be kept under regular surveillance.
On demand treatment
Patients on on-demand treatment should be instructed to contact their physician if their symptoms change in character.
Helicobacter pylori eradication
When prescribing Esomeprazole for eradication of Helicobacter pylori possible drug interactions for all components in the triple therapy should be considered. Clarithromycin is a potent inhibitor of CYP3A4 and hence contraindications and interactions for Clarithromycin should be considered when the triple therapy is used in patients concurrently taking other medicinal products metabolised via CYP3A4 such as Cisapride.
Gastrointestinal infections
Treatment with proton pump inhibitors may lead to slightly increased risk of gastrointestinal infections such as Salmonella and Campylobacter.
Absorption of Vitamin B12
Esomeprazole, as all acid-blocking medicines, may reduce the absorption of Vitamin B12 (Cyanocobalamin) due to hypo- or achlorhydria. This should be considered in patients with reduced body stores or risk factors for reduced Vitamin B12 absorption on long-term therapy.
Hypomagnesaemia
Severe hypomagnesaemia has been reported in patients treated with proton pump inhibitors (PPIs) like Esomeprazole for at least three months, and in most cases for a year. Serious manifestations of hypomagnesaemia such as fatigue, tetany, delirium, convulsions, dizziness and ventricular arrhythmia can occur but they may begin insidiously and be overlooked. In most affected patients, hypomagnesaemia improved after Magnesium replacement and discontinuation of the PPI.
For patients expected to be on prolonged treatment or who take PPIs with Digoxin or medicinal products that may cause hypomagnesaemia (e.g. diuretics), health care professionals should consider measuring magnesium levels before starting PPI treatment and periodically during treatment.
Risk of fracture
Proton pump inhibitors, especially if used in high doses and over long durations (> 1 year), may modestly increase the risk of hip, wrist and spine fracture, predominantly in the elderly or in presence of other recognised risk factors. Observational studies suggest that proton pump inhibitors may increase the overall risk of fracture by 10 - 40%. Some of this increase may be due to other risk factors. Patients at risk of osteoporosis should receive care according to current clinical guidelines and they should have an adequate intake of Vitamin D and Calcium.
Subacute cutaneous lupus erythematosus (SCLE)
Proton pump inhibitors are associated with very infrequent cases of SCLE. If lesions occur, especially in sunexposed areas of the skin, and if accompanied by arthralgia, the patient should seek medical help promptly and the health care professional should consider stopping Esomeprazole. SCLE after previous treatment with a proton pump inhibitor may increase the risk of SCLE with other proton pump inhibitors.
Combination with other medicinal products
Co-administration of Esomeprazole with Atazanavir is not recommended. If the combination of Atazanavir with a proton pump inhibitor is judged unavoidable, close clinical monitoring is recommended in combination with an increase in the dose of Atazanavir to 400 mg with 100 mg of Ritonavir; Esomeprazole 20 mg should not be exceeded. Esomeprazole is a CYP2C19 inhibitor. When starting or ending treatment with Esomeprazole, the potential for interactions with medicinal products metabolised through CYP2C19 should be considered. An interaction is observed between Clopidogrel and Esomeprazole. The clinical relevance of this interaction is uncertain. As a precaution, concomitant use of Esomeprazole and Clopidogrel should be discouraged.
When prescribing Esomeprazole for on demand therapy, the implications for interactions with other pharmaceuticals, due to fluctuating plasma concentrations of Esomeprazole should be considered.
Interference with laboratory tests
Increased Chromogranin A (CgA) level may interfere with investigations for neuroendocrine tumours. To avoid this interference, Esomeprazole treatment should be stopped for at least 5 days before CgA measurements. If CgA and gastrin levels have not returned to reference range after initial measurement, measurements should be repeated 14 days after cessation of proton pump inhibitor treatment.
4.6 Drugs interactions
Effects of Esomeprazole on the pharmacokinetics of other medicinal products
Medicinal products with pH dependent absorption
Gastric acid suppression during treatment with Esomeprazole and other PPIs might decrease or increase the absorption of medicinal products with a gastric pH dependent absorption. As with other medicinal products that decrease intra-gastric acidity, the absorption of medicinal products such as Ketoconazole, Itraconazole and Erlotinib can decrease and the absorption of Digoxin can increase during treatment with Esomeprazole. Concomitant treatment with Omeprazole (20 mg daily) and Digoxin in healthy subjects increased the bioavailability of Digoxin by 10% (up to 30% in two out of ten subjects). Digoxin toxicity has been rarely reported. However, caution should be exercised when Esomeprazole is given at high doses in elderly patients. Therapeutic drug monitoring of Digoxin should then be reinforced.
Protease inhibitors
Omeprazole has been reported to interact with some protease inhibitors. The clinical importance and the mechanisms behind these reported interactions are not always known. Increased gastric pH during Omeprazole treatment may change the absorption of the protease inhibitors. Other possible interaction mechanisms are via inhibition of CYP 2C19.
For Atazanavir and Nelfinavir, decreased serum levels have been reported when given together with Omeprazole and concomitant administration is not recommended. Co-administration of Omeprazole (40 mg once daily) with Atazanavir 300 mg / Ritonavir 100 mg to healthy volunteers resulted in a substantial reduction in Atazanavir exposure (approximately 75% decrease in AUC, Cmax and Cmin). Increasing the Atazanavir dose to 400 mg did not compensate for the impact of Omeprazole on Atazanavir exposure. The co-administration of Omeprazole (20 mg qd) with Atazanavir 400 mg / Ritonavir 100 mg to healthy volunteers resulted in a decrease of approximately 30% in the Atazanavir exposure as compared with the exposure observed with Atazanavir 300 mg / Ritonavir 100 mg qd without Omeprazole 20 mg qd. Co-administration of Omeprazole (40 mg qd) reduced mean Nelnavir AUC, Cmax and Cmin by 36 - 39% and mean AUC, Cmax and Cmin for the pharmacologically active metabolite M8 was reduced by 75 - 92%. Due to the similar pharmacodynamic effects and pharmacokinetic properties of Omeprazole and Esomeprazole, concomitant administration with Esomeprazole and Atazanavir is not recommended and concomitant administration with Esomeprazole and Nelnavir is contraindicated.
For Saquinavir (with concomitant Ritonavir), increased serum levels (80 - 100%) have been reported during concomitant Omeprazole treatment (40 mg qd). Treatment with Omeprazole 20 mg qd had no effect on the exposure of Darunavir (with concomitant Ritonavir) and Amprenavir (with concomitant Ritonavir). Treatment with Esomeprazole 20 mg qd had no effect on the exposure of Amprenavir (with and without concomitant Ritonavir). Treatment with Omeprazole 40 mg qd had no effect on the exposure of Lopinavir (with concomitant Ritonavir).
Methotrexate
When given together with PPIs, Methotrexate levels have been reported to increase in some patients. In high-dose Methotrexate administration, a temporary withdrawal of Esomeprazole may need to be considered.
Tacrolimus
Concomitant administration of Esomeprazole has been reported to increase the serum levels of Tacrolimus. A reinforced monitoring of tacrolimus concentrations as well as renal function (creatinine clearance) should be performed, and dosage of tacrolimus adjusted, if needed.
Medicinal products metabolised by CYP2C19
Esomeprazole inhibits CYP2C19, the major Esomeprazole-metabolising enzyme. Thus, when Esomeprazole is combined with medicinal products metabolised by CYP2C19, such as Diazepam, Citalopram, Imipramine, Clomipramine, Phenytoin, etc., the plasma concentrations of these medicinal products may be increased and a dose reduction could be needed. This should be considered especially when prescribing Esomeprazole for on-demand therapy.
Diazepam
Concomitant administration of 30 mg Esomeprazole resulted in a 45% decrease in clearance of the CYP2C19 substrate Diazepam.
Phenytoin
Concomitant administration of 40 mg Esomeprazole resulted in a 13% increase in trough plasma levels of Phenytoin in epileptic patients. It is recommended to monitor the plasma concentrations of phenytoin when treatment with Esomeprazole is introduced or withdrawn.
Voriconazole
Omeprazole (40 mg once daily) increased Voriconazole (a CYP2C19 substrate) Cmax and AUC by 15% and 41% respectively.
Warfarin
Concomitant administration of 40 mg Esomeprazole to Warfarin-treated patients in a clinical trial showed that coagulation times were within the accepted range. However, post-marketing, a few isolated cases of elevated INR of clinical significance have been reported during concomitant treatment. Monitoring is recommended when initiating and ending concomitant Esomeprazole treatment during treatment with warfarin or other Coumarine derivatives.
Cilostazol
Omeprazole as well as Esomeprazole act as inhibitors of CYP2C19. Omeprazole, given in doses of 40 mg to healthy subjects in a cross-over study, increased Cmax and AUC for Cilostazol by 18% and 26% respectively and one of its active metabolites by 29% and 69% respectively.
Cisapride
In healthy volunteers, concomitant administration of 40 mg Esomeprazole resulted in a 32% increase in area under the plasma concentration-time curve (AUC) and a 31% prolongation of elimination half-life (t½) but no significant increase in peak plasma levels of Cisapride. The slightly prolonged QTc interval observed after administration of Cisapride alone, was not further prolonged when Cisapride was given in combination with Esomeprazole.
Clopidogrel
Results from studies in healthy subjects have shown a pharmacokinetic (PK) / pharmacodynamic (PD) interaction between Clopidogrel (300 mg loading dose / 75 mg daily maintenance dose) and Esomeprazole (40 mg p.o. daily) resulting in decreased exposure to the active metabolite of Clopidogrel by an average of 40% and resulting in decreased maximum inhibition of (ADP induced) platelet aggregation by an average of 14%.
Investigated medicinal products with no clinically relevant interaction
Amoxicillin and Quinidine
Esomeprazole has been shown to have no clinically relevant effects on the pharmacokinetics of amoxicillin or Quinidine.
Naproxen or Rofecoxib
Studies evaluating concomitant administration of Esomeprazole and either naproxen or Rofecoxib did not identify any clinically relevant pharmacokinetic interactions during short-term studies.
Effects of other medicinal products on the pharmacokinetics of Esomeprazole
Medicinal products which inhibit CYP2C19 and/or CYP3A4
Esomeprazole is metabolised by CYP2C19 and CYP3A4. Concomitant administration of Esomeprazole and a CYP3A4 inhibitor, Clarithromycin (500 mg b.i.d.), resulted in a doubling of the exposure (AUC) to Esomeprazole. Concomitant administration of Esomeprazole and a combined inhibitor of CYP2C19 and CYP3A4 may result in more than doubling of the Esomeprazole exposure. The CYP2C19 and CYP3A4 inhibitor Voriconazole increased
Omeprazole AUC by 280 %. A dose adjustment of Esomeprazole is not regularly required in either of these situations. However, dose adjustment should be considered in patients with severe hepatic impairment and if long-term treatment is indicated.
Medicinal products which induce CYP2C19 and/or CYP3A4
Medicinal products known to induce CYP2C19 or CYP3A4 or both (such as Rifampicin and St John's wort) may lead to decreased Esomeprazole serum levels by increasing the Esomeprazole metabolism.
Paediatric population
Interaction studies have only been performed in adults.
4.7 Fertility, pregnancy and lactation
Fertility
Animal studies with the racemic mixture Omeprazole, given by oral administration do not indicate effects with respect to fertility.
Pregnancy
Clinical data on exposed pregnancies with Esomeprazole are insufficient. With the racemic mixture, Omeprazole, data on a larger number of exposed pregnancies stemmed from epidemiological studies indicate no malformative nor foeto-toxic effects. Animal studies with Esomeprazole do not indicate direct or indirect harmful effects with respect to embryonal / foetal development. Animal studies with the racemic mixture do not indicate direct or indirect harmful effects with respect to pregnancy, parturition or postnatal development. Caution should be exercised when prescribing to pregnant women.
A moderate amount of data on pregnant women (between 300 - 1000 pregnancy outcomes) indicates no malformative or foeto/neonatal toxicity of Esomeprazole.
Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity.
Breast-feeding
It is not known whether Esomeprazole is excreted in human breast milk. There is insufficient information on the effects of Esomeprazole in new-borns / infants. Esomeprazole should not be used during breast-feeding.
4.7 Effects on ability to drive and use machines
(uncommon) and blurred vision (rare) has been reported. If affected patients should not drive or use machines. Undesirable effects
Summary of the safety profile
Headache, abdominal pain, diarrhoea and nausea are among those adverse reactions that have been most commonly reported in clinical trials (and also from post-marketing use). In addition, the safety profile is similar for different formulations, treatment indications, age groups and patient populations. No dose-related adverse reactions have been identified.
Tabulated list of adverse reactions
The following adverse drug reactions have been identified or suspected in the clinical trials programme for Esomeprazole and post-marketing. None was found to be dose-related. The reactions are classified according to frequency Very common ≥ 1/10; Common ≥ 1/100 to < 1/10; Uncommon ≥ 1/1,000 to < 1/100; Rare ≥ 1/10,000 to < 1/1,000; Very rare < 1/10,000; Not known (cannot be estimated from the available data).
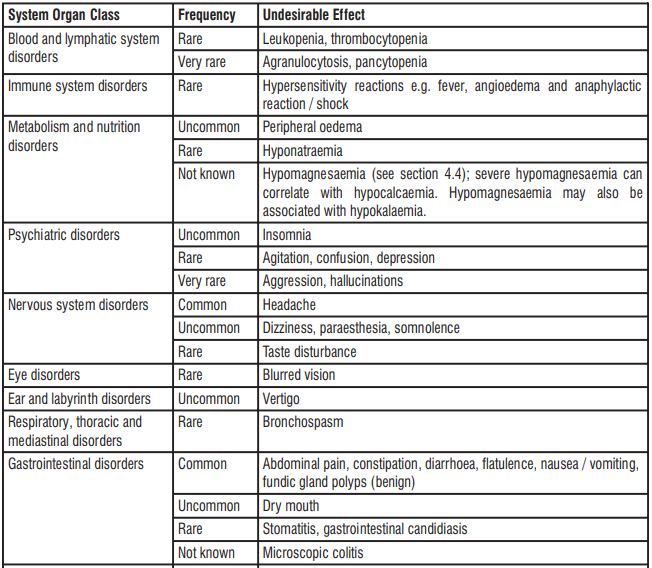
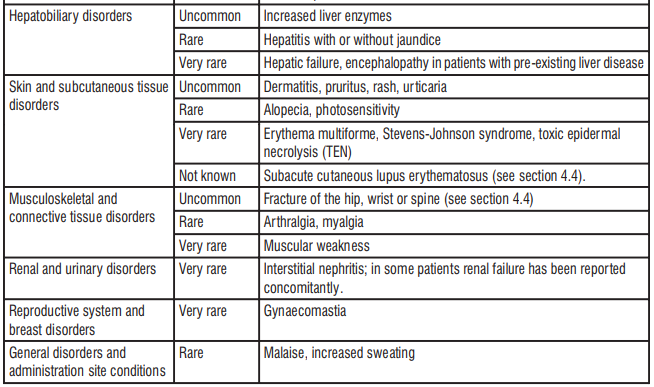
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to :medico@zuventus.com
Website : https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is very limited experience to date with deliberate overdose.
Symptoms
The symptoms described in connection with 280 mg were gastrointestinal symptoms and weakness. Single doses of 80 mg Esomeprazole were uneventful.
Management
No specific antidote is known. Esomeprazole is extensively plasma protein bound and is therefore not readily dialyzable. As in any case of overdose, treatment should be symptomatic and general supportive measures should be utilised.
5.0 Pharmacological properties
5.1 Mechanism of action
Esomeprazole is a weak base and is concentrated and converted to the active form in the highly acidic environment of the secretory canaliculi of the parietal cell, where it inhibits the enzyme H+/K+-ATPase – the acid pump and inhibits both basal and stimulated acid secretion.
5.2 Pharmacodynamic properties
Esomeprazole is the S-isomer of Omeprazole and reduces gastric acid secretion through a specific targeted mechanism of action. It is a specific inhibitor of the acid pump in the parietal cell. Both the R- and S-isomer of Omeprazole have similar pharmacodynamic activity.
Pharmacodynamic effects
After oral dosing with Esomeprazole 20 mg and 40 mg the onset of effect occurs within one hour. After repeated administration with 20 mg Esomeprazole once daily for five days, mean peak acid output after pentagastrin stimulation is decreased 90% when measured 6 - 7 hours after dosing on day five. After five days of oral dosing with 20 mg and 40 mg of Esomeprazole, intragastric pH above 4 was maintained for a mean time of 13 hours and 17 hours, respectively over 24 hours in symptomatic GERD patients. The proportion of patients maintaining an intragastric pH above 4 for at least 8, 12 and 16 hours respectively were for Esomeprazole 20 mg 76%, 54% and 24%. Corresponding proportions for Esomeprazole 40 mg were 97%, 92% and 56%. Using AUC as a surrogate parameter for plasma concentration, a relationship between inhibition of acid secretion and exposure has been shown.
Healing of reflux esophagitis with Esomeprazole 40 mg occurs in approximately 78% of patients after four weeks, and in 93% after eight weeks. One-week treatment with Esomeprazole 20 mg b.i.d. and appropriate antibiotics, results in successful eradication of H. pylori in approximately 90% of patients. After eradication treatment for one week there is no need for subsequent monotherapy with antisecretory medicinal products for effective ulcer healing and symptom resolution in uncomplicated duodenal ulcers. During treatment with antisecretory medicinal products serum gastrin increases in response to the decreased acid secretion. Also CgA increases due to decreased gastric acidity. The increased CgA level may interfere with investigations for neuroendocrine tumours. Available published evidence suggests that proton pump inhibitors should be discontinued between 5 days and 2 weeks prior to CgA measurements. This is to allow CgA levels that might be spuriously elevated following PPI treatment to return to reference range. An increased number of ECL cells possibly related to the increased serum gastrin levels, have been observed in both children and adults during long term treatment with Esomeprazole. The findings are considered to be of no clinical significance.
5.3 Pharmacokinetic properties
Absorption
Esomeprazole is acid labile and is administered orally as gastro-resistant granules. In vivo conversion to the R-isomer is negligible. Absorption of Esomeprazole is rapid, with peak plasma levels occurring approximately 1 - 2 hours after dose. The absolute bioavailability is 64% after a single dose of 40 mg and increases to 89% after repeated once daily administration. For 20 mg Esomeprazole, the corresponding values are 50% and 68%, respectively
Food intake both delays and decreases the absorption of Esomeprazole although this has no significant influence on the effect of Esomeprazole on intragastric acidity.
Distribution
The apparent volume of distribution at steady state in healthy subjects is approximately 0.22 l/kg body weight. Esomeprazole is 97% plasma protein bound.
Biotransformation
Esomeprazole is completely metabolised by the cytochrome P450 system (CYP). The major part of the metabolism of Esomeprazole is dependent on the polymorphic CYP2C19, responsible for the formation of the hydroxy- and desmethyl metabolites of Esomeprazole. The remaining part is dependent on another specific isoform, CYP3A4, responsible for the formation of Esomeprazole sulphone, the main metabolite in plasma.
Elimination
The parameters below reflect mainly the pharmacokinetics in individuals with a functional CYP2C19 enzyme, extensive metabolisers.
Total plasma clearance is about 17 l/h after a single dose and about 9 l/h after repeated administration. The plasma elimination half-life is about 1.3 hours after repeated once daily dosing. Esomeprazole is completely eliminated from plasma between doses with no tendency for accumulation during once-daily administration. The major metabolites of Esomeprazole have no effect on gastric acid secretion. Almost 80% of an oral dose of Esomeprazole is excreted as metabolites in the urine, the remainder in the faeces. Less than 1% of the parent drug is found in urine.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development. Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows : No new or unexpected toxicity findings were observed in juvenile rats and dogs, after administration of Esomeprazole for up to 3 months, as compared to adult animals. Carcinogenicity studies in the rat with the racemic mixture have shown gastric ECL-cell hyperplasia and carcinoids. These gastric effects in the rat are the result of sustained, pronounced hypergastrinaemia secondary to reduced production of gastric acid and are observed after long-term treatment in the rat with inhibitors of gastric acid secretion.
No new or unexpected toxicity ndings were observed in juvenile rats and dogs, after administration of Esomeprazole for up to 3 months, as compared to adult animals.
7.0 Description
Zosa FAST contains Esomeprazole Magnesium Trihydrate (with Sodium Bicarbonate as buffer). It is a proton pump inhibitor and decreases gastric acid secretion.
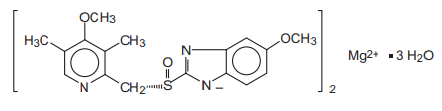
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Alu-Alu blister strip of 10 tablets.
8.4 Storage and handing instructions Store protected from moisture, at a temperature not exceeding 30°C.
Keep out of reach of children.
9.0 Patient Counselling Information
Acute tubulointerstitial nephritis
Advise the patient or caregiver to call the patient’s healthcare provider immediately if they experience signs and/or symptoms associated with suspected acute TIN.
Clostridium difficile-associated diarrhea
Advise the patient or caregiver to immediately call the patient’s healthcare provider if they experience diarrhea that does not improve.
Bone fracture
Advise the patient or caregiver to report any fractures, especially of the hip, wrist or spine, to the patient’s healthcare provider.
Severe cutaneous adverse reactions
Advise the patient or caregiver to discontinue Zosa FAST and immediately call the patient’s healthcare provider for at first appearance of a severe cutaneous adverse reaction or other sign of hypersensitivity signs or symptoms associated with Severe Cutaneous Adverse Reactions.
Cutaneous and systemic lupus erythematosus
Advise the patient or caregiver to immediately call the patient’s healthcare provider for any new or worsening of symptoms associated with cutaneous or systemic lupus erythematosus.
Cyanocobalamin (Vitamin B12) deficiency
Advise the patient or caregiver to report any clinical symptoms that may be associated with Cyanocobalamin deficiency to the patient’s healthcare provider if they have been receiving Zosa FAST for longer than 3 years.
Hypomagnesemia and mineral metabolism
Advise the patient or caregiver to report any clinical symptoms that may be associated with hypomagnesemia, hypocalcemia, and/or hypokalemia to the patient’s healthcare provider, if they have been receiving Zosa FAST for at least 3 months.
Drug interactions
Advise the patient or caregiver to report to their healthcare provider if starting treatment with Rilpivirine-containing products, Clopidogrel, St. John’s Wort or Rifampin or if they take high-dose Methotrexate.
Administration
- Take Zosa FAST tablet at least one hour before meals.
- Antacids may be used concomitantly with Zosa FAST.
- Swallow Zosa FAST tablet whole; do not chew or crush the tablet.
12.0 Date of issue
16 November 2023
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
1. What Esomeprazole is and what it is used for
2. What you need to know before you take Esomeprazole
3. How to take Esomeprazole
4. Possible side effects
5. How to store Esomeprazole
6. Contents of the pack and other information
1. What Esomeprazole is and what it is used for
Esomeprazole contains a medicine called esomeprazole. This belongs to a group of medicines called ‘proton pump inhibitors’. They work by reducing the amount of acid that your stomach produces.
Esomeprazole is used to treat the following conditions:
Adults
- Gastroesophageal reflux disease’ (GERD). This is where acid from the stomach escapes into the gullet (the tube which connects your throat to your stomach) causing pain, inflammation and heartburn.
- Ulcers in the stomach or upper part of the gut (intestine) that are infected with bacteria called ‘Helicobacter pylori’. If you have this condition, your doctor may also prescribe antibiotics to treat the infection and allow the ulcer to heal. Stomach ulcers caused by medicines called NSAIDs (Non-Steroidal Anti-Inflammatory Drugs). Esomeprazole can also be used to stop stomach ulcers or ulcers in the upper part of the gut (intestine) from forming if you are taking NSAIDs.
- Too much acid in the stomach caused by a growth in the pancreas (Zollinger-Ellison syndrome).
- Prolonged treatment after prevention of rebleeding of ulcers with intravenous esomeprazole.
Adolescents aged 12 years and above
- ‘Gastroesophageal reflux disease’ (GERD). This is where acid from the stomach escapes into the gullet (the tube which connects your throat to your stomach) causing pain, inflammation and heartburn.
- Ulcers in the stomach or upper part of the gut (intestine) that are infected with bacteria called ‘Helicobacter pylori’. If you have this condition, your doctor may also prescribe antibiotics to treat the infection and allow the ulcer to heal.
2. What you need to know before you take Esomeprazole
Do not take Esomeprazole
- If you are allergic to esomeprazole or any of the other ingredients of this medicine.
- If you are allergic to other proton pump inhibitor medicines (e.g. pantoprazole, lansoprazole, rabeprazole, omeprazole).
- If you are taking a medicine containing nelfinavir (used to treat HIV infection).
Do not take Esomeprazole if any of the above applies to you. If you are not sure, talk to your doctor or pharmacist before taking Esomeprazole.
Warnings and precautions
Talk to your doctor or pharmacist before taking Esomeprazole:
- If you have severe liver problems.
- If you have severe kidney problems.
- If you have a vitamin B12 deficiency or there is a risk of a vitamin B12 deficiency.
- If you have ever had a skin reaction after treatment with a medicine similar to Esomeprazole that reduces stomach acid.
- If you are due to have a specific blood test (Chromogranin A).
Esomeprazole may hide the symptoms of other diseases. Therefore, if any of the following happen to you before you start taking Esomeprazole or while you are taking it, talk to your doctor straight away:
- You lose a lot of weight for no reason and have problems swallowing.
- You get stomach pain or indigestion.
- You begin to vomit food or blood.
- You pass black stools (blood-stained faeces).
If you have been prescribed Esomeprazole “on demand” you should contact your doctor if your symptoms continue or change in character.
Taking a proton pump inhibitor like Esomeprazole, especially over a period of more than one year, may slightly increase your risk of fracture in the hip, wrist or spine. Tell your doctor if you have osteoporosis or if you are taking corticosteroids (which can increase the risk of osteoporosis).
Rash and skin symptoms
If you get a rash on your skin, especially in areas exposed to the sun tell your doctor as soon as you can, as you may need to stop your treatment with Esomeprazole. Remember to also mention any other ill-effects like pain in your joints.
Serious skin rashes have occurred in patients taking esomeprazole. The rash can involve ulcers of the mouth, throat, nose, genitals and conjunctivitis (red and swollen eyes). These serious skin rashes often come after flu-like symptoms such as fever, headache, body ache. The rash may cover large parts of the body with blistering and peeling of the skin. If at any time during the treatment (even after several weeks) you develop a rash or any of these skin symptoms, stop taking this medicine and contact your doctor immediately.
Children under the age of 12 years
Information on dosing for children aged 1 to 11 years is provided in the product information of other dosage forms e.g. sachet (ask your doctor or pharmacist if you require further information).
Other medicines and Esomeprazole
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines. This is because Esomeprazole can affect the way some medicines work and some medicines can have an effect on Esomeprazole.
Do not take Esomeprazole if you are taking a medicine containing nelfinavir (used to treat HIV infection).
Tell your doctor or pharmacist if you are taking any of the following medicines:
- Atazanavir (used to treat HIV infection).
- Clopidogrel (used to prevent blood clots).
- Ketoconazole, itraconazole or voriconazole (used to treat infections caused by a fungus).
- Erlotinib (used to treat cancer).
- Citalopram, imipramine or clomipramine (used to treat depression).
- Diazepam (used to treat anxiety, relax muscles or in epilepsy).
- Phenytoin (used in epilepsy). If you are taking phenytoin, your doctor will need to monitor you when you start or stop taking Esomeprazole.
- Medicines that are used to thin your blood, such as warfarin. Your doctor may need to monitor you when you start or stop taking Esomeprazole.
- Cilostazol (used to treat intermittent claudication – a pain in your legs when you walk which is caused by an insufficient blood supply).
- Cisapride (used for indigestion and heartburn).
- Digoxin (used for heart problems).
- Methotrexate (a chemotherapy medicine used in high doses to treat cancer) – if you are taking a high dose of methotrexate, your doctor may temporarily stop your Esomeprazole treatment.
- Tacrolimus (organ transplantation).
- Rifampicin (used for treatment of tuberculosis).
- St. John’s wort (Hypericum perforatum) (used to treat depression).
If your doctor has prescribed the antibiotics amoxicillin and clarithromycin as well as Esomeprazole to treat ulcers caused by Helicobacter pylori infection, it is very important that you tell your doctor about any other medicines you are taking.
Esomeprazole with food and drink
You can take the tablets with food or on an empty stomach.
Pregnancy, breast-feeding and fertility
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Your doctor will decide whether you can take Esomeprazole during this time. It is not known if Esomeprazole passes into breast milk.
Therefore, you should not take Esomeprazole if you are breast-feeding. Driving and using machines Esomeprazole is not likely to affect you being able to drive or use any tools or machines.
However, side effects such as dizziness and blurred vision may uncommonly or rarely occur. If affected, you should not drive or use machines.
3. How to take Esomeprazole
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
- If you are taking this medicine for a long time, your doctor will want to monitor you (particularly if you are taking it for more than a year).
- If your doctor has told you to take this medicine as and when you need it, tell your doctor if your symptoms change.
How much to take
- Your doctor will tell you how many tablets to take and how long to take them for. This will depend on your condition, how old you are and how well your liver works.
- The usual doses are given below.
Use in adults aged 18 and above
To treat heartburn caused by gastroesophageal reflux disease (GERD):
- If your doctor has found that your food pipe (gullet) has been slightly damaged, the recommended dose is one Esomeprazole 40 mg tablet once a day for 4 weeks. Your doctor may tell you to take the same dose for a further 4 weeks if your gullet has not yet healed.
- The recommended dose once the gullet has healed is one Esomeprazole 20 mg tablet once a day.
- If your gullet has not been damaged, the recommended dose is one Esomeprazole 20 mg tablet each day. Once the condition has been controlled, your doctor may tell you to take your medicine as and when you need it, up to a maximum of one Esomeprazole 20 mg tablet each day.
- If you have severe liver problems, your doctor may give you a lower dose.
To treat ulcers caused by Helicobacter pylori infection and to stop them coming back
- The recommended dose is one Esomeprazole 20 mg tablet twice a day for one week.
- Your doctor will also tell you to take antibiotics for example amoxicillin and clarithromycin. To treat stomach ulcers caused by NSAIDs (Non-Steroidal Anti-Inflammatory Drugs)
- The recommended dose is one Esomeprazole 20 mg tablet once a day for 4 to 8 weeks. To prevent stomach ulcers if you are taking NSAIDs (Non-Steroidal Anti-Inflammatory Drugs)
- The recommended dose is one Esomeprazole 20 mg tablet once a day. To treat too much acid in the stomach caused by a growth in the pancreas (Zollinger-Ellison syndrome)
- The recommended dose is Esomeprazole 40 mg twice a day.
- Your doctor will adjust the dose depending on your needs and will also decide how long you need to take the medicine for. The maximum dose is 80 mg twice a day.
Prolonged treatment after prevention of rebleeding of ulcers with intravenous esomeprazole
The recommended dose is one Esomeprazole 40 mg tablet once a day for 4 weeks.
Use in adolescents aged 12 or above
To treat heartburn caused by gastroesophageal reflux disease (GERD)
- If your doctor has found that your food pipe (gullet) has been slightly damaged, the recommended dose is one Esomeprazole 40 mg gastro-resistant tablet once a day for 4 weeks. Your doctor may tell you to take the same dose for a further 4 weeks if your gullet has not yet healed.
- The recommended dose once the gullet has healed is one Esomeprazole 20 mg gastro-resistant tablet once a day.
- If your gullet has not been damaged, the recommended dose is one Esomeprazole 20 mg gastro-resistant tablet each day.
- If you have severe liver problems, your doctor may give you a lower dose.
- Swallow the tablets whole with a drink of water. Do not chew or crush the tablets. This is because the tablets contain coated pellets which stop the medicine from being broken down by the acid in your stomach. It is important not to damage the pellets.
What to do if you have trouble swallowing the tablets
- If you have trouble swallowing the tablets:
- Put them into a glass of still (non-fizzy) water. Do not use any other liquids.
- Stir until the tablets break up (the mixture will not be clear). Then drink the mixture straight away or within 30 minutes. Always stir the mixture just before drinking it.
- To make sure that you have drunk all of the medicine, rinse the glass very well with half a glass of water and drink it. The solid pieces contain the medicine - do not chew or crush them.
- If you cannot swallow at all, the tablet can be mixed with some water and put into a syringe. It can then be given to you through a tube directly into your stomach (‘gastric tube’).
Use in children under the age of 12 years
Esomeprazole gastro-resistant tablets are not recommended for children less than 12 years old.
Elderly
Dose adjustment is not required in the elderly. If you take more Esomeprazole than you should If you take more Esomeprazole than prescribed by your doctor, talk to your doctor or pharmacist straight away.
If you forget to take Esomeprazole
- If you forget to take a dose, take it as soon as you remember it. However, if it is almost time for your next dose, skip the missed dose.
- Do not take a double dose (two doses at the same time) to make up for a forgotten dose.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
If you notice any of the following serious side effects, stop taking Esomeprazole and contact a doctor immediately:
- Yellow skin, dark urine and tiredness which can be symptoms of liver problems. These effects are rare and may affect up to 1 in 1,000 people.
- Sudden wheezing, swelling of your lips, tongue and throat or body, rash, fainting or difficulties in swallowing (severe allergic reaction). These effects are rare and may affect up to 1 in 1,000 people.
- Sudden onset of a severe rash or reddening of the skin with blisters or peeling may occur even after several weeks of treatment. There may also be severe blisters and bleeding in the lips, eyes, mouth, nose and genitals. The skin rashes may develop into serious widespread skin damage (peeling of the epidermis and superficial mucous membranes) with life threatening consequences. This could be ‘erythema multiforme’, ‘Stevens-Johnson syndrome’, ‘toxic epidermal necrolysis’ or ‘drug reaction with eosinophilia and systemic symptoms’. These effects are very rare and might affect up to 1 in 10,000 people.
Other side effects include:
Common (may affect up to 1 in 10 people)
- Headache.
- Effects on your stomach or gut: diarrhoea, stomach pain, constipation, wind (flatulence).
- Feeling sick (nausea) or being sick (vomiting).
- Benign polyps in the stomach.
Uncommon (may affect up to 1 in 100 people)
- Swelling of the feet and ankles.
- Disturbed sleep (insomnia).
- Dizziness, tingling feelings such as “pins and needles”, feeling sleepy.
- Spinning feeling (vertigo).
- Dry mouth.
- Changes in blood tests that check how the liver is working.
- Skin rash, lumpy rash (hives) and itchy skin.
- Fracture of the hip, wrist or spine (if Esomeprazole is used in high doses and over long duration).
Rare (may affect up to 1 in 1,000 people)
- Blood problems such as a reduced number of white cells or platelets. This can cause weakness, bruising or make infections more likely.
- Low levels of sodium in the blood. This may cause weakness, being sick (vomiting) and cramps.
- Feeling agitated, confused or depressed.
- Taste changes.
- Eyesight problems such as blurred vision.
- Suddenly feeling wheezy or short of breath (bronchospasm).
- An inflammation of the inside of the mouth.
- An infection called “thrush” which can affect the gut and is caused by a fungus.
- Liver problems, including jaundice which can cause yellow skin, dark urine, and tiredness.
- Hair loss (alopecia).
- Skin rash on exposure to sunshine.
- Joint pains (arthralgia) or muscle pains (myalgia).
- Generally feeling unwell and lacking energy.
- Increased sweating.
Very rare (may affect up to 1 in 10,000 people)
- Changes in blood count including agranulocytosis (lack of white blood cells).
- Aggression.
- Seeing, feeling or hearing things that are not there (hallucinations).
- Severe liver problems leading to liver failure and inflammation of the brain.
- Sudden onset of a severe rash or blistering or peeling skin. This may be associated with a high fever and joint pains (Erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms).
- Muscle weakness.
- Severe kidney problems.
- Enlarged breasts in men.
Not known (frequency cannot be estimated from the available data)
- If you are on esomeprazole for more than three months it is possible that the levels of magnesium in your blood may fall. Low levels of magnesium can be seen as fatigue, involuntary muscle contractions, disorientation, convulsions, dizziness or increased heart rate. If you get any of these symptoms, please tell your doctor promptly. Low levels of magnesium can also lead to a reduction in potassium or calcium levels in the blood. Your doctor may decide to perform regular blood tests to monitor your levels of magnesium.
- Inflammation in the gut (leading to diarrhoea).
- Rash, possibly with pain in the joints.
Esomeprazole may in very rare cases affect the white blood cells leading to immune deficiency. If you have an infection with symptoms such as fever with a severely reduced general condition or fever with symptoms of a local infection such as pain in the neck, throat or mouth or difficulties in urinating, you must consult your doctor as soon as possible so that a lack of white blood cells (agranulocytosis) can be ruled out by a blood test. It is important for you to give information about your medication at this time.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top of the home page. By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Esomeprazole
- Keep this medicine out of the sight and reach of children.
- Do not use this medicine after the expiry date which is stated on the blister and the carton after [Exp]. The expiry date refers to the last day of that month.
- Store below 30°C.
- Store in the original package in order to protect from moisture.
- Do not throw away medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
Each film coated tablet contains:
Esomeprazole Magnesium Trihydrate IP equivalent to Esomeprazole 40 mg
(with Sodium Bicarbonate as buffer)
Revised on 09/24
For More Information About This Product
Ursomax 450 Tablets
1.0 Generic name
Ursodeoxycholic acid
2.0 Qualitative and quantitative composition
Ursomax 150
Each uncoated tablet contains
Ursodeoxycholic acid IP.…………………………150 mg
Excipients……………………………………………...…...q.s.
Ursomax 300
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………300 mg
Excipients……………………………………………...…...q.s.
Ursomax 450 SR
Each prolonged-release uncoated tablet contains
Ursodeoxycholic acid IP.……………………………450 mg
Excipients……………………………………………......q.s.
3.0 Dosage form and strength
Tablet 150 mg/300 mg/450 mg
4.0 Clinical particulars
4.1 Therapeutic Indication
For the dissolution of radiolucent cholesterol gallstone, chronic cholestatic liver diseases in particular primary biliary cirrhosis, primary sclerosing cholangitis & cholestasis associated with cystic fibrosis.
4.2 Posology and method of administration
The dosage should be calculated based on the patient's body weight. The calculated dosage should be rounded to the nearest number of tablets.
Dissolving of cholesterol stones
Usual dosage: 8 to 10 mg/kg/day, corresponding to, for example, four to six 150 mg tablets, or two to three tablets of 300 mg, or two tablets of 450 mg. The daily dose can be administered two or three times after the meals: two tablets should always be taken after the evening meal.
Also a single evening dose can be selected (e.g., a patient of 60 kg, in the evening two tablets of 300 mg). Preferably, this single dose should be taken one hour before bedtime and ± two hours after the evening meal with a glass of milk or a small snack.
The duration of the treatment in order to obtain lysis of the gallstones depends on their size, but is usually not shorter than three to four months. To assess the result of the therapy properly, it is necessary to determine the size of the stones accurately at the start of the treatment and to check this further regularly, for example every six months, by means of a new contrast X-ray recording and/or sonographic recording.
In patients in whom, after six months of treatment with the indicated dosage, the stones are not reduced in size, it is recommended to determine the lithogenic index in the bile by means of a duodenum drainage. When the bile has an index of > 1.0, it is unlikely that a favourable result can be obtained, and it is better to consider a different form of treatment for the gallstones.
Treatment should be continued for three to four months after it is established by means of ultrasound check that the gallstones are completely dissolved. An interruption of the treatment for three to four weeks results in a return to over-saturation of the bile and prolongs the overall duration of the therapy. The interruption of the treatment after the dissolving of the gallstones can be followed by a recurrence.
Primary Biliary Cholangitis (Primary biliary cirrhosis)
The dosage of Ursodeoxycholic acid in primary biliary cholangitis (stages I-III), amounts to 12-15 mg/kg/day, which is equivalent to four to eight tablets of 150 mg, two to four tablets of 300 mg, to be taken in two to three portions during the day, or with two tablets of 450 mg, to be taken in two portions during the day.
The dosage of ursodeoxycholic acid in primary biliary cholangitis stage IV and an increase of the serum bilirubin contents (> 40 μg/l), should be in the first instance, only a half of the normal dose (6 to 8 mg/kg/day). Thereafter, the liver function should be closely monitored for several weeks (once every two weeks for six weeks). If there is no deterioration of the liver function (AF, ALT (SGPT), AST (SGOT), γ-GT, bilirubin) and no increase in itching occurs, the dose may be further increased to the usual level. Moreover, the liver function must then again be closely monitored for several weeks. If again no deterioration of the liver function takes place, the patient may be held at the normal dosage for a long time.
In patients with primary biliary cholangitis stage IV without elevated serum bilirubin, the usual starting dose is allowed to be administered directly. Anyway, here too an accurate control of the liver function should be executed.
The treatment of the primary biliary cholangitis should be regularly assessed on the basis of liver values (laboratory) and clinical findings.
Paediatric population
Children with cystic fibrosis aged 6 years to 18 years
Treatment of hepatobiliary diseases as a result of cystic fibrosis 20 mg/kg/day divided in two to three divided doses. If necessary, increase to 30 mg/kg/day. This corresponds with four to ten tablets of 150mg, two to five tablets of 300 mg, or with two to three tablets of 450 mg, to be taken in one or two portions during the day
Method of administration
If the patient has difficulty in swallowing because of the size of the tablet, the tablet can be halved if necessary on the dividing score, so that one half tablet can be taken twice directly in sequence.
4.3 Contraindications
Ursodeoxycholic acid tablets should not be used in patients with:
- Acute inflammation of the gall bladder or bile ducts
- Occlusion of the biliary tract (occlusion of the common bile duct or a cystic duct)
- Frequent episodes of biliary colic
- X-ray radiolucent calcified gallstones
- Impaired contractility of the gallbladder
- Hypersensitivity to bile acids or to any of the excipient
- Active gastric and duodenal ulcers
Paediatric population
- Unsuccessful portoenterostomy or without recovery of good bile flow in children with biliary atresia.
4.4 Special warnings and precautions for use
Ursodeoxycholic acid tablets should be taken under medical supervision.
During the first three months of the treatment liver function parameters AST (SGOT), ALT (SGPT) and γ-GT should be monitored by the physician every 4 weeks, thereafter every 3 months. Apart from allowing for identification of responders and non-responders in patients being treated for primary biliary cholangitis, this monitoring would also enable an early detection of potential hepatic deterioration, particularly in patients with advanced primary biliary cholangitis.
When used for dissolving gallstones:
In order to be able to assess the therapeutic progression of the dissolution of gallstones and to timely identify a possible calcification of the stones, the gall bladder, depending on the size of the stones, should be visualized 6 to 10 months after the start of the treatment (oral cholecystography) with total image and occlusions and in the standing and lying position (ultrasound investigation).
If the gallbladder cannot be visualized on X-rays, or in cases of calcified gallstones, impaired contractility of the gall bladder or frequent episodes of biliary colic, the treatment with Ursodeoxycholic acid should be discontinued.
When used for the treatment of advanced primary biliary cholangitis:
In very rare cases decompensation of liver cirrhosis is observed which partially decreased after treatment discontinuation.
In patients with PBC, the clinical symptoms may worsen in rare cases at the start of treatment, e.g. pruritus may increase. In this case, the therapy is to be continued with a dose reduction and subsequently should be gradually increased to the recommended dose as described in section 4.2.
If diarrhoea occurs, the dosage should be reduced, and treatment should be discontinued in case of persistent diarrhoea.
Female patients who use Ursodeoxycholic acid for dissolving gall stones must use an effective non-hormonal method of contraception, since hormonal contraception may increase biliary lithiasis
4.5 Drugs interactions
- Ursodeoxycholic acid tablets should not be used concurrently with colestyramine, colestipol, or an antacid, on the basis of aluminium hydroxide and/or smectite (aluminium oxide), because these preparations bind ursodeoxycholic acid in the intestine and thereby inhibits its absorption and efficacy. If the use of such a medicine is necessary, must it be taken at least 2 hours before or after ursomax.
- Ursodeoxycholic acid may affect the absorption of ciclosporin from the intestine. In patients treated with ciclosporin the blood level of ciclosporin should be monitored and the ciclosporin dose should be adjusted, if necessary.
- In isolated cases Ursodeoxycholic acid can reduce the absorption of ciprofloxacin.
- In a clinical study in healthy volunteers, the concomitant use of UDCA (500 mg/day) and rosuvastatin (20 mg/day) resulted in slightly elevated plasma levels of rosuvastatin.The clinical relevance of this interaction, also with other statins, is not known.
- Ursodeoxycholic acid has been shown to reduce the peak plasma concentration (Cmax) and the AUC of the calcium antagonist nitrendipine in healthy volunteers. Close monitoring of the outcome of concurrent use of nitrendipine and ursodeoxycholic acid is recommended. An increase of the dose of nitrendipine may be necessary. An interaction with a reduction of the therapeutic effect of dapsone was also reported. These observations, together with in vitro findings could be an indication that ursodeoxycholic acid can induce cytochrome P450 3A enzymes. Induction has, however not been observed in a well-designed interaction study with budesonide, which is a known cytochrome P450 3A substrate.
- Oestrogens and blood cholesterol lowering agents such as clofibrate increase hepatic cholesterol secretion and may therefore encourage biliary lithiasis; which is a counter-effect to ursodeoxycholic acid used for dissolution of gallstones.
4.6 Use in special populations
Pregnancy
- There are no or limited amount of data from the use of ursodeoxycholic acid in pregnant women. Studies in animals have shown reproductive toxicity during the early gestation phase.
- Ursodeoxycholic acid must not be used during pregnancy, unless clearly necessary.
Women of childbearing potentialc
- Women of childbearing potential should be treated with ursodeoxycholic acid, only if they practice reliable contraception: non-hormonal contraceptives or oral contraceptives with low oestrogen dose are recommended. However, in patients taking Ursodeoxycholic acid for dissolving gallstones an effective non-hormonal contraception should be used, since hormonal oral contraceptives may increase biliary lithiasis.
- The possibility of a pregnancy must be excluded before beginning treatment.
Nursing Mothers
- According to few documented cases of breastfeeding women milk levels of ursodeoxycholic acid levels in milk are very low and probably no adverse reactions are to be expected in breastfed infants.
Fertility
Animal studies did not show an influence of ursodeoxycholic acid on fertility. Human data on fertility treatment with ursodeoxycholic acid are not available.
4.7 Effects on ability to drive and use machines
Ursodeoxycholic acid has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
The following adverse reactions have been reported during clinical trials and are ranked using the following frequency:
very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data).
Gastrointestinal disorders:
In clinical studies, reports of pasty stools or diarrhoea during treatment with ursodeoxycholic acid were common.
In very rare cases, severe right upper abdominal pain has occurred during the treatment of primary biliary cholangitis.
Hepatobiliary disorders:
During treatment with ursodeoxycholic acid calcification of gallstones can occur in very rare cases.
During the treatment of advanced stages of primary biliary cholangitis decompensation of cirrhosis has been observed in very rare cases, which partially regressed after treatment discontinuation.
Hypersensitivity reactions:
Very rarely urticaria may occur.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
4.9 Overdose
In the case of overdose diarrhoea may occur. In general, other symptoms of overdose are unlikely, because the absorption of the ursodeoxycholic acid decreases with increasing dose and therefore more is excreted in the faeces.
If diarrhoea occurs, the dosage should be reduced, and treatment should be discontinued in case of persistent diarrhoea. No specific measures are needed and the consequences of diarrhoea should be treated symptomatically with restoration of fluid and electrolyte balance.
Additional information or special populations: Long-term, high-dose UDCA therapy (28-30 mg/kg/day) by patients with primary sclerosing cholangitis (off-label use) was associated with a higher frequency of serious adverse events.
5.0 Pharmacological properties
5.1 Mechanism of Action
The ursodeoxycholic acid converts lithogenic bile in non-lithogenic bile and gradually dissolves the cholesterol gallstones.
5.2 Pharmacodynamic properties
Bile acids are among the most important components of the bile and play a role in the stimulation of bile secretion. Bile acids are also important to keep the cholesterol in bile in solution. In a healthy person, the ratio between the concentration of cholesterol and bile acids in the bile is such that the cholesterol will remain in solution for most of the day. In this case, no gallstones can form (the bile is non-lithogenic). In patients with cholesterol stones in the bile, this ratio is changed and the bile is supersaturated with cholesterol (bile is lithogenic). This may cause a precipitation of cholesterol crystals and the formation of gallstones after some time.
The ursodeoxycholic acid converts lithogenic bile in non-lithogenic bile and gradually dissolves the cholesterol gallstones.
Investigations of the effect of ursodeoxycholic acid on the cholestasis in patients with impaired biliary drainage and on the clinical symptoms in patients with primary biliary cholangitis and cystic fibrosis have shown that cholestatic symptoms in the blood (to be measured by the increased value of alkaline phosphatase (AF), gamma-GT and bilirubin) and the itch declined rapidly, while also the fatigue decreased in the majority of patients. Moreover, studies seem to indicate a positive benefit-risk ratio of the ursodeoxycholic acid in children and young adult cystic fibrosis patients with mild to moderate hepatobiliary disorders.
Paediatric population
Cystic fibrosis
From clinical reports long-term experience of 10 years and more has been gained with UDCA therapy in paediatric patients suffering from cystic fibrosis associated hepatobiliary disorders (CFAHD). There is evidence that treatment with UDCA can inhibit bile duct proliferation, can halt progression of histological damage and even reverse hepato-biliary changes, if it happens at an early stage of CFAHD. The treatment with UDCA should be started as soon as the CFAHD diagnosis is made, in order to optimize the effectiveness of the treatment.
5.3 Pharmacokinetic properties
About 90% of the therapeutic dose of the ursodeoxycholic acid is rapidly absorbed in the small intestine after oral administration.
After the absorption, ursodeoxycholic acid is absorbed in the liver (there is a substantial "first-passeffect"), where it is conjugated with glycine or taurine and then secreted into the bile ducts. Only a small portion of ursodeoxycholic acid is found in the systemic circulation. This is excreted renally. With the exception of conjugation, ursodeoxycholic acid is not metabolised. However, a small fraction of orally administered ursodeoxycholic acid undergoes bacterial conversion to 7-keto-lithocholic acid resp. lithocholic acid after each enterohepatic circulation, while bacterial deconjugation also takes place in the duodenum. Ursodeoxycholic acid, 7-keto-lithocholic acid and lithocholic acid are relatively poorly soluble in water, so a large part of it is excreted via the bile into the faeces. Resorbed ursodeoxycholic acid is conjugated again by the liver; 80% of the lithocholic acid formed in the duodenum is excreted in the faeces, but the remaining 20% of it are sulphated by the liver to insoluble lithocholylconjugates after absorption, which in turn are excreted via the bile and faeces. Resorbed 7- keto-lithocholic acid is reduced to chenodeoxycholic acid in the liver. Lithocholic acid can cause cholestatic liver damage, when the liver is unable to sulphate the lithocholic acid. Although a reduced capacity to sulphate the lithocholic acid in the liver is found in some patients, there is for the time being no clinical evidence that cholestatic liver damage can be associated with the therapy using ursodeoxycholic acid.
After repeated dosage, the ursodeoxycholic acid concentration in the bile reaches a "steady state" after approximately 3 weeks: the total concentration of the ursodeoxycholic acid, however, is never higher than about 60% of the total concentration of the bile acid in the bile: also at high doses.
After therapy with ursodeoxycholic acid is stopped, the concentration of ursodeoxycholic acid in bile decreases quickly after 1 week to 5-10% of the "steady-state" concentration.
The biological half-life of ursodeoxycholic acid is approximately 3.5 to 5.8 days.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Acute toxicity
Acute toxicity studies in animals have not revealed any toxic damage.
b) Chronic toxicity
Subchronic toxicity studies in monkeys showed hepatotoxic effects in the groups given high doses, including functional changes (e.g. liver enzyme changes) and morphological changes such as bile duct proliferation, portal inflammatory foci and hepatocellular necrosis. These toxic effects are most likely attributable to lithocholic acid, a metabolite of ursodeoxycholic acid, which in monkeys – unlike humans – is not detoxified. Clinical experience confirms that the described hepatotoxic effects are of no apparent relevance in humans.
c) Carcinogenic and mutagenic potential
Long-term studies in mice and rats revealed no evidence of ursodeoxycholic acid having carcinogenic potential. In vitro and in vivo genetic toxicology tests with ursodeoxycholic acid were negative. The tests with ursodeoxycholic acid revealed no relevant evidence of a mutagenic effect.
d) Toxicity to reproduction
In studies in rats, tail malformations occurred after a dose of 2000 mg per kg of body weight. In rabbits, no teratogenic effects were found, although there were embryotoxic effects (from a dose of 100 mg per kg of body weight). ursodeoxycholic acid had no effect on fertility in rats and did not affect peri-/post-natal development of the offspring.
7.0 Description
Ursodeoxycholic acid (UDCA) is a naturally occurring bile acid found in small quantities in normal human bile and in larger quantities in the biles of certain species of bears. The chemical name of ursodiol is 3α,7ß-dihydroxy-5ß-cholan-24-oic (C24H40O4)
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on pack
8.3 Packaging information
10 Blister strips of 10 tablets each
8.4 Storage and handing instructions
- Store in a cool, dry & dark place.
- Keep out of reach of children.
- Tablet should be swallowed whole & not to be broken, chewed or crushed.
9.0 Patient counselling information
- Ursomax tablet should be taken after a meal with a glass of milk or water.
- Eat a healthy diet, exercise regularly, and avoid alcohol intake.
- Diarrhea may occur as a side effect. Drink plenty of fluids and inform your doctor if diarrhea persists or if you find blood in your stools.
- Your doctor may monitor your liver function and bilirubin levels every month for the next 3 months after the start of therapy, and every 6 months thereafter.
- Do not stop taking the medication without talking to your doctor.
12.0 Date of Revision
28.08.2024
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
1. What URSOMAX is and what it is used for
2. What you need to know before you take URSOMAX
3. How to take URSOMAX
4. Possible side effects
5. How to store URSOMAX
6. Contents of the pack and other information
1. WHAT URSOMAX IS AND WHAT IT IS USED FOR
The active ingredient in Ursomax tablet is Ursodeoxycholic Acid, which is a naturally occurring substance (bile acid) normally present in the bile.
Ursodeoxycholic Acid influences the composition of the bile, by which cholesterol gallstones can be solved. The effects of Ursodeoxycholic Acid in primary biliary cholangitis and cystic fibrosis can be explained by various mechanisms, such as a protective effect on the liver cells and an effect on the immune system.
1. URSOMAX Tablets is used in patients:
- with small gallstones
- who do not want to undergo surgery or are not eligible for gallstone surgery
- in whom too much cholesterol has been found in the bile
2. URSOMAX Tablets is used in patients with primary biliary cholangitis. Primary biliary cholangitis is a disease in which liver tissue is damaged by an impaired drainage of the bile.
3. URSOMAX Tablets is used in children aged 6 years to 18 years old with liver and biliary diseases caused by cystic fibrosis. Cystic fibrosis, also referred to as mucoviscidosis, is an inherited disorder in which the mucus is particularly tough in the whole body. This may cause, among other conditions, clogging and inflammation in the liver and in the bile ducts.
2. WHAT YOU NEED TO KNOW BEFORE YOU TAKE URSOMAX
Do not use URSOMAX
- You have an acute inflammation of the gallbladder or the bile ducts.
- You have a narrowing or blockage of the bile ducts.
- You suffer from often occurring cramp-like pain in the upper abdomen (biliary colic).
- You have calcified gallstones that do not transmit X-rays.
- You have a gall bladder that cannot properly constrict any more.
- You are allergic to bile acids or any of the other ingredients of this medicine.
- You have an active gastric or duodenal ulcer.
Children
In children with disrupted biliary drainage due to production of connective tissue in the bile duct (biliary atresia) in whom the bile flow is not restored by healing or by an artificial bile duct (portoenterostomy).
Warnings and precautions
This medicine must be used under medical supervision.
Your doctor should examine your liver every 4 weeks during the first three months of treatment. Then this should be done every 3 months. Apart from allowing for identification of responders and non-responders in patients being treated for primary biliary cholangitis, this monitoring would also enable an early detection of potential hepatic deterioration, particularly in patients with advanced primary biliary cholangitis.
When used for dissolving gallstones:
In order to be able to assess the therapeutic progression of the dissolution of gallstones and to timely identify a possible calcification of the stones, the gall bladder, depending on the size of the stones, should be visualized 6 to 10 months after the start of the treatment (oral cholecystography) with total image and occlusions and in the standing and lying position (ultrasound control).
If the gallbladder cannot be visualized on X-rays, or in cases of calcified gallstones, impaired contractility of the gall bladder or frequent episodes of biliary colic, the treatment with Ursodeoxycholic Tablets should be discontinued.
When used for the treatment of advanced primary biliary cholangitis:
In very rare cases decompensation of liver cirrhosis is observed which partially decreased after treatment discontinuation. Women who use URSOMAX for dissolving gallstones should stop using the birth control pill and other methods used to prevent pregnancy, because the hormones in the birth control pill can promote the production of gallstones.
When you are in the final stage of primary biliary cholangitis it can in very rare cases occur that your liver function is strongly reduced. The liver function will partly recover after stopping the treatment.
If you experience problems with diarrhoea, your doctor will reduce the dose. If the diarrhoea persists, your doctor may decide to stop the treatment.
Contact your doctor or pharmacist before you start to use this medicine.
Other medicines and URSOMAX Tablets
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines. This also applies for medicines obtained without a prescription.
The effect of the following drugs can be influenced (interactions):
A reduced effect of the following medicines is possible when using this medicine:
- Medicines binding stomach acid based on aluminium hydroxide and bile acid binding substances (colestyramine, colestipol) can bind the ursodeoxycholic acid and thereby prevent its absorption. Therefore, these medicines may not be taken simultaneously with URSOMAX, but always 2 hours before or after.
- Ursodeoxycholic acid can reduce the absorption of ciprofloxacin, dapsone (antibiotics) and nitrendipine (antihypertensive agent) from the intestine. When one of these resources has been used simultaneously with URSOMAX, your doctor will carefully supervise you.
An intensified effect of the following medicines is possible when using this medicine:
- URSOMAX may increase the absorption of cyclosporine from the intestine: if necessary, the dosing guided by the cyclosporine concentration should be adjusted in the blood.
Oestrogens, oral contraceptives ("the pill") and cholesterol-lowering agents (such as clofibrate) may promote the forming of gallstones, and can thus counteract the effect of the treatment of gallstones with URSOMAX Tablets
Inform your doctor or pharmacist, if you are taking or have recently taken other medicines. This also applies for medicines obtained without a prescription.
Pregnancy and breast-feeding
If you are pregnant or breastfeeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Pregnancy:
You should not use this medicine during pregnancy unless your doctor considers it absolutely necessary.
Women of child-bearing potential
Even if you are not pregnant, you should discuss this possibility with your doctor, because women of childbearing age may only be treated, if they use a reliable method of contraception. Non-hormonal contraception or contraception with low dose oestrogens is recommended. However, if you use this medicine for solving gallstones, you may only use non-hormonal contraception, because hormonal contraception promotes the formation of gallstones.
Breast-feeding
Tell your doctor if you are breastfeeding or about to start breastfeeding. According to few documented cases of breastfeeding women milk levels of ursodeoxycholic acid in milk are very low and probably no adverse reactions are to be expected in breastfed infants.
Driving and using machines
URSOMAX has no or negligible influence on driving ability or the ability to operate machines.
3. HOW TO USE URSOMAX
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
Your doctor will determine your dose based on your body weight.
Take the tablets after a meal with a glass of milk or a small snack. Take the prescribed number of tablets distributed throughout the day.
1. Dissolving of gallstones:
Four to six tablets of 150 mg, two to three tablets of 300 mg or two tablets of 450 mg per day (600- 900 mg ursodeoxycholic acid per day).
Two tablets should always be taken after dinner.
When taking a dose two or three times daily: for example, one tablet after lunch and two tablets after dinner.
When taking one dose of two tablets daily: take both tablets preferably two hours after dinner one hour before going to sleep.
2. Primary biliary cholangitis (damage to liver tissue by impaired bile flow):
- Phase I-III: four to eight tablets of 150 mg, two to four tablets of 300 mg, or two tablets of 450 mg per day (600-1200 mg ursodeoxycholic acid per day).
Take the prescribed dose in two to three servings per day after meals.
- Phase IV: On the basis of liver function examination your doctor will determine if a normal dosage, as in phase I-III, or the half of this dose will be prescribed to you.
Children with cystic fibrosis aged 6 to 18 years
3. Disorders of liver and biliary tract caused by cystic fibrosis (mucoviscidosis):
four to ten tablets of 150 mg, two to five tablets of 300 mg, or two to three tablets of 450 mg per day (600-1500 mg ursodeoxycholic acid per day).
The tablets with a score can be divided if you have problems in swallowing because of the size of the tablets, so that one half tablet can be taken twice directly in sequence.
If you notice that Ursodeoxycholic Acid Tablets is too strong or on the contrary too weak, consult your doctor or pharmacist.
If you take more URSOMAX than you should
You should tell your doctor if you have taken more Ursodeoxycholic Acid Tablets than you should. It is unlikely that you will notice any problems but you may experience diarrhoea.
If you forget to take URSOMAX
Take the prescribed amount at the next regular taking time. Do not take a double dose to make up for a forgotten dose.
How long should you use URSOMAX Tablets?
The duration of the treatment depends on the size of the gallstone, but is usually not shorter than three to four months. Treatment should not be interrupted prematurely; even if the symptoms have disappeared.
Only an X-ray or an ultrasound scan can show that the gallstones are completely dissolved. After it has been shown with the aid of an echogram that the gallstones have disappeared completely, the treatment should still be continued for three to four months.
The use of Ursodeoxycholic Acid in the treatment of primary biliary cholangitis and disorders of the liver and the biliary system as a result of cystic fibrosis will usually be maintained continuously, in order to continue to maintain the protective effect of Ursodeoxycholic Acid.
Do you have any further questions on the use of this medicine? Please contact your doctor or pharmacist.
4. POSSIBLE SIDE EFFECTS
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Common side effects (occurring in fewer than 1 in 10 patients but more than 1 in 100 patients)
- pasty stools or diarrhoea
Very rare side effects (may affect up to 1 in 10,000 people)
- in the treatment of primary biliary cholangitis: severe pain in the right upper abdomen, severe deterioration (decompensation) of the liver cirrhosis which partially decreases after cessation of treatment;
- calcification of gallstones;
- urticaria (hives)
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. HOW TO STORE URSOMAX
- Store in a cool, dry & dark place.
- Keep out of reach of children.
- Tablet should be swallowed whole & not to be broken, chewed or crushed.
6. CONTENTS OF THE PACK AND OTHER INFORMATION
What URSOMAX contains
Ursomax 150
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………150 mg
Excipients……………………………………………..…...q.s.
Ursomax 300
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………300 mg
Excipients……………………………………………..…...q.s.
Ursomax 450 SR
Each prolonged-release uncoated tablet contains
Ursodeoxycholic acid IP.……………………………450 mg
Excipients……………………………………………..…...q.s.
What URSOMAX looks like and contents of the pack
10 Blister strips of 10 tablets each
Marketing authorisation holder and manufacturer
Zuventus Healthcare Ltd
Kamerey Bhasmay, Elaka Pakyong,
Rangpo, East-Sikkim 737 132.
Registered Trade Mark of Zuventus.
This leaflet was last revised in 08/2023
For More Information About This Product
Ursomax 300 Tablets
1.0 Generic name
Ursodeoxycholic acid
2.0 Qualitative and quantitative composition
Ursomax 150
Each uncoated tablet contains
Ursodeoxycholic acid IP.…………………………150 mg
Excipients……………………………………………...…...q.s.
Ursomax 300
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………300 mg
Excipients……………………………………………...…...q.s.
Ursomax 450 SR
Each prolonged-release uncoated tablet contains
Ursodeoxycholic acid IP.……………………………450 mg
Excipients……………………………………………......q.s.
3.0 Dosage form and strength
Tablet 150 mg/300 mg/450 mg
4.0 Clinical particulars
4.1 Therapeutic Indication
For the dissolution of radiolucent cholesterol gallstone, chronic cholestatic liver diseases in particular primary biliary cirrhosis, primary sclerosing cholangitis & cholestasis associated with cystic fibrosis.
4.2 Posology and method of administration
The dosage should be calculated based on the patient's body weight. The calculated dosage should be rounded to the nearest number of tablets.
Dissolving of cholesterol stones
Usual dosage: 8 to 10 mg/kg/day, corresponding to, for example, four to six 150 mg tablets, or two to three tablets of 300 mg, or two tablets of 450 mg. The daily dose can be administered two or three times after the meals: two tablets should always be taken after the evening meal.
Also a single evening dose can be selected (e.g., a patient of 60 kg, in the evening two tablets of 300 mg). Preferably, this single dose should be taken one hour before bedtime and ± two hours after the evening meal with a glass of milk or a small snack.
The duration of the treatment in order to obtain lysis of the gallstones depends on their size, but is usually not shorter than three to four months. To assess the result of the therapy properly, it is necessary to determine the size of the stones accurately at the start of the treatment and to check this further regularly, for example every six months, by means of a new contrast X-ray recording and/or sonographic recording.
In patients in whom, after six months of treatment with the indicated dosage, the stones are not reduced in size, it is recommended to determine the lithogenic index in the bile by means of a duodenum drainage. When the bile has an index of > 1.0, it is unlikely that a favourable result can be obtained, and it is better to consider a different form of treatment for the gallstones.
Treatment should be continued for three to four months after it is established by means of ultrasound check that the gallstones are completely dissolved. An interruption of the treatment for three to four weeks results in a return to over-saturation of the bile and prolongs the overall duration of the therapy. The interruption of the treatment after the dissolving of the gallstones can be followed by a recurrence.
Primary Biliary Cholangitis (Primary biliary cirrhosis)
The dosage of Ursodeoxycholic acid in primary biliary cholangitis (stages I-III), amounts to 12-15 mg/kg/day, which is equivalent to four to eight tablets of 150 mg, two to four tablets of 300 mg, to be taken in two to three portions during the day, or with two tablets of 450 mg, to be taken in two portions during the day.
The dosage of ursodeoxycholic acid in primary biliary cholangitis stage IV and an increase of the serum bilirubin contents (> 40 μg/l), should be in the first instance, only a half of the normal dose (6 to 8 mg/kg/day). Thereafter, the liver function should be closely monitored for several weeks (once every two weeks for six weeks). If there is no deterioration of the liver function (AF, ALT (SGPT), AST (SGOT), γ-GT, bilirubin) and no increase in itching occurs, the dose may be further increased to the usual level. Moreover, the liver function must then again be closely monitored for several weeks. If again no deterioration of the liver function takes place, the patient may be held at the normal dosage for a long time.
In patients with primary biliary cholangitis stage IV without elevated serum bilirubin, the usual starting dose is allowed to be administered directly. Anyway, here too an accurate control of the liver function should be executed.
The treatment of the primary biliary cholangitis should be regularly assessed on the basis of liver values (laboratory) and clinical findings.
Paediatric population
Children with cystic fibrosis aged 6 years to 18 years
Treatment of hepatobiliary diseases as a result of cystic fibrosis 20 mg/kg/day divided in two to three divided doses. If necessary, increase to 30 mg/kg/day. This corresponds with four to ten tablets of 150mg, two to five tablets of 300 mg, or with two to three tablets of 450 mg, to be taken in one or two portions during the day
Method of administration
If the patient has difficulty in swallowing because of the size of the tablet, the tablet can be halved if necessary on the dividing score, so that one half tablet can be taken twice directly in sequence.
4.3 Contraindications
Ursodeoxycholic acid tablets should not be used in patients with:
- Acute inflammation of the gall bladder or bile ducts
- Occlusion of the biliary tract (occlusion of the common bile duct or a cystic duct)
- Frequent episodes of biliary colic
- X-ray radiolucent calcified gallstones
- Impaired contractility of the gallbladder
- Hypersensitivity to bile acids or to any of the excipient
- Active gastric and duodenal ulcers
Paediatric population
- Unsuccessful portoenterostomy or without recovery of good bile flow in children with biliary atresia.
4.4 Special warnings and precautions for use
Ursodeoxycholic acid tablets should be taken under medical supervision.
During the first three months of the treatment liver function parameters AST (SGOT), ALT (SGPT) and γ-GT should be monitored by the physician every 4 weeks, thereafter every 3 months. Apart from allowing for identification of responders and non-responders in patients being treated for primary biliary cholangitis, this monitoring would also enable an early detection of potential hepatic deterioration, particularly in patients with advanced primary biliary cholangitis.
When used for dissolving gallstones:
In order to be able to assess the therapeutic progression of the dissolution of gallstones and to timely identify a possible calcification of the stones, the gall bladder, depending on the size of the stones, should be visualized 6 to 10 months after the start of the treatment (oral cholecystography) with total image and occlusions and in the standing and lying position (ultrasound investigation).
If the gallbladder cannot be visualized on X-rays, or in cases of calcified gallstones, impaired contractility of the gall bladder or frequent episodes of biliary colic, the treatment with Ursodeoxycholic acid should be discontinued.
When used for the treatment of advanced primary biliary cholangitis:
In very rare cases decompensation of liver cirrhosis is observed which partially decreased after treatment discontinuation.
In patients with PBC, the clinical symptoms may worsen in rare cases at the start of treatment, e.g. pruritus may increase. In this case, the therapy is to be continued with a dose reduction and subsequently should be gradually increased to the recommended dose as described in section 4.2.
If diarrhoea occurs, the dosage should be reduced, and treatment should be discontinued in case of persistent diarrhoea.
Female patients who use Ursodeoxycholic acid for dissolving gall stones must use an effective non-hormonal method of contraception, since hormonal contraception may increase biliary lithiasis
4.5 Drugs interactions
- Ursodeoxycholic acid tablets should not be used concurrently with colestyramine, colestipol, or an antacid, on the basis of aluminium hydroxide and/or smectite (aluminium oxide), because these preparations bind ursodeoxycholic acid in the intestine and thereby inhibits its absorption and efficacy. If the use of such a medicine is necessary, must it be taken at least 2 hours before or after ursomax.
- Ursodeoxycholic acid may affect the absorption of ciclosporin from the intestine. In patients treated with ciclosporin the blood level of ciclosporin should be monitored and the ciclosporin dose should be adjusted, if necessary.
- In isolated cases Ursodeoxycholic acid can reduce the absorption of ciprofloxacin.
- In a clinical study in healthy volunteers, the concomitant use of UDCA (500 mg/day) and rosuvastatin (20 mg/day) resulted in slightly elevated plasma levels of rosuvastatin.The clinical relevance of this interaction, also with other statins, is not known.
- Ursodeoxycholic acid has been shown to reduce the peak plasma concentration (Cmax) and the AUC of the calcium antagonist nitrendipine in healthy volunteers. Close monitoring of the outcome of concurrent use of nitrendipine and ursodeoxycholic acid is recommended. An increase of the dose of nitrendipine may be necessary. An interaction with a reduction of the therapeutic effect of dapsone was also reported. These observations, together with in vitro findings could be an indication that ursodeoxycholic acid can induce cytochrome P450 3A enzymes. Induction has, however not been observed in a well-designed interaction study with budesonide, which is a known cytochrome P450 3A substrate.
- Oestrogens and blood cholesterol lowering agents such as clofibrate increase hepatic cholesterol secretion and may therefore encourage biliary lithiasis; which is a counter-effect to ursodeoxycholic acid used for dissolution of gallstones.
4.6 Use in special populations
Pregnancy
- There are no or limited amount of data from the use of ursodeoxycholic acid in pregnant women. Studies in animals have shown reproductive toxicity during the early gestation phase.
- Ursodeoxycholic acid must not be used during pregnancy, unless clearly necessary.
Women of childbearing potentialc
- Women of childbearing potential should be treated with ursodeoxycholic acid, only if they practice reliable contraception: non-hormonal contraceptives or oral contraceptives with low oestrogen dose are recommended. However, in patients taking Ursodeoxycholic acid for dissolving gallstones an effective non-hormonal contraception should be used, since hormonal oral contraceptives may increase biliary lithiasis.
- The possibility of a pregnancy must be excluded before beginning treatment.
Nursing Mothers
- According to few documented cases of breastfeeding women milk levels of ursodeoxycholic acid levels in milk are very low and probably no adverse reactions are to be expected in breastfed infants.
Fertility
Animal studies did not show an influence of ursodeoxycholic acid on fertility. Human data on fertility treatment with ursodeoxycholic acid are not available.
4.7 Effects on ability to drive and use machines
Ursodeoxycholic acid has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
The following adverse reactions have been reported during clinical trials and are ranked using the following frequency:
very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data).
Gastrointestinal disorders:
In clinical studies, reports of pasty stools or diarrhoea during treatment with ursodeoxycholic acid were common.
In very rare cases, severe right upper abdominal pain has occurred during the treatment of primary biliary cholangitis.
Hepatobiliary disorders:
During treatment with ursodeoxycholic acid calcification of gallstones can occur in very rare cases.
During the treatment of advanced stages of primary biliary cholangitis decompensation of cirrhosis has been observed in very rare cases, which partially regressed after treatment discontinuation.
Hypersensitivity reactions:
Very rarely urticaria may occur.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
4.9 Overdose
In the case of overdose diarrhoea may occur. In general, other symptoms of overdose are unlikely, because the absorption of the ursodeoxycholic acid decreases with increasing dose and therefore more is excreted in the faeces.
If diarrhoea occurs, the dosage should be reduced, and treatment should be discontinued in case of persistent diarrhoea. No specific measures are needed and the consequences of diarrhoea should be treated symptomatically with restoration of fluid and electrolyte balance.
Additional information or special populations: Long-term, high-dose UDCA therapy (28-30 mg/kg/day) by patients with primary sclerosing cholangitis (off-label use) was associated with a higher frequency of serious adverse events.
5.0 Pharmacological properties
5.1 Mechanism of Action
The ursodeoxycholic acid converts lithogenic bile in non-lithogenic bile and gradually dissolves the cholesterol gallstones.
5.2 Pharmacodynamic properties
Bile acids are among the most important components of the bile and play a role in the stimulation of bile secretion. Bile acids are also important to keep the cholesterol in bile in solution. In a healthy person, the ratio between the concentration of cholesterol and bile acids in the bile is such that the cholesterol will remain in solution for most of the day. In this case, no gallstones can form (the bile is non-lithogenic). In patients with cholesterol stones in the bile, this ratio is changed and the bile is supersaturated with cholesterol (bile is lithogenic). This may cause a precipitation of cholesterol crystals and the formation of gallstones after some time.
The ursodeoxycholic acid converts lithogenic bile in non-lithogenic bile and gradually dissolves the cholesterol gallstones.
Investigations of the effect of ursodeoxycholic acid on the cholestasis in patients with impaired biliary drainage and on the clinical symptoms in patients with primary biliary cholangitis and cystic fibrosis have shown that cholestatic symptoms in the blood (to be measured by the increased value of alkaline phosphatase (AF), gamma-GT and bilirubin) and the itch declined rapidly, while also the fatigue decreased in the majority of patients. Moreover, studies seem to indicate a positive benefit-risk ratio of the ursodeoxycholic acid in children and young adult cystic fibrosis patients with mild to moderate hepatobiliary disorders.
Paediatric population
Cystic fibrosis
From clinical reports long-term experience of 10 years and more has been gained with UDCA therapy in paediatric patients suffering from cystic fibrosis associated hepatobiliary disorders (CFAHD). There is evidence that treatment with UDCA can inhibit bile duct proliferation, can halt progression of histological damage and even reverse hepato-biliary changes, if it happens at an early stage of CFAHD. The treatment with UDCA should be started as soon as the CFAHD diagnosis is made, in order to optimize the effectiveness of the treatment.
5.3 Pharmacokinetic properties
About 90% of the therapeutic dose of the ursodeoxycholic acid is rapidly absorbed in the small intestine after oral administration.
After the absorption, ursodeoxycholic acid is absorbed in the liver (there is a substantial "first-passeffect"), where it is conjugated with glycine or taurine and then secreted into the bile ducts. Only a small portion of ursodeoxycholic acid is found in the systemic circulation. This is excreted renally. With the exception of conjugation, ursodeoxycholic acid is not metabolised. However, a small fraction of orally administered ursodeoxycholic acid undergoes bacterial conversion to 7-keto-lithocholic acid resp. lithocholic acid after each enterohepatic circulation, while bacterial deconjugation also takes place in the duodenum. Ursodeoxycholic acid, 7-keto-lithocholic acid and lithocholic acid are relatively poorly soluble in water, so a large part of it is excreted via the bile into the faeces. Resorbed ursodeoxycholic acid is conjugated again by the liver; 80% of the lithocholic acid formed in the duodenum is excreted in the faeces, but the remaining 20% of it are sulphated by the liver to insoluble lithocholylconjugates after absorption, which in turn are excreted via the bile and faeces. Resorbed 7- keto-lithocholic acid is reduced to chenodeoxycholic acid in the liver. Lithocholic acid can cause cholestatic liver damage, when the liver is unable to sulphate the lithocholic acid. Although a reduced capacity to sulphate the lithocholic acid in the liver is found in some patients, there is for the time being no clinical evidence that cholestatic liver damage can be associated with the therapy using ursodeoxycholic acid.
After repeated dosage, the ursodeoxycholic acid concentration in the bile reaches a "steady state" after approximately 3 weeks: the total concentration of the ursodeoxycholic acid, however, is never higher than about 60% of the total concentration of the bile acid in the bile: also at high doses.
After therapy with ursodeoxycholic acid is stopped, the concentration of ursodeoxycholic acid in bile decreases quickly after 1 week to 5-10% of the "steady-state" concentration.
The biological half-life of ursodeoxycholic acid is approximately 3.5 to 5.8 days.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Acute toxicity
Acute toxicity studies in animals have not revealed any toxic damage.
b) Chronic toxicity
Subchronic toxicity studies in monkeys showed hepatotoxic effects in the groups given high doses, including functional changes (e.g. liver enzyme changes) and morphological changes such as bile duct proliferation, portal inflammatory foci and hepatocellular necrosis. These toxic effects are most likely attributable to lithocholic acid, a metabolite of ursodeoxycholic acid, which in monkeys – unlike humans – is not detoxified. Clinical experience confirms that the described hepatotoxic effects are of no apparent relevance in humans.
c) Carcinogenic and mutagenic potential
Long-term studies in mice and rats revealed no evidence of ursodeoxycholic acid having carcinogenic potential. In vitro and in vivo genetic toxicology tests with ursodeoxycholic acid were negative. The tests with ursodeoxycholic acid revealed no relevant evidence of a mutagenic effect.
d) Toxicity to reproduction
In studies in rats, tail malformations occurred after a dose of 2000 mg per kg of body weight. In rabbits, no teratogenic effects were found, although there were embryotoxic effects (from a dose of 100 mg per kg of body weight). ursodeoxycholic acid had no effect on fertility in rats and did not affect peri-/post-natal development of the offspring.
7.0 Description
Ursodeoxycholic acid (UDCA) is a naturally occurring bile acid found in small quantities in normal human bile and in larger quantities in the biles of certain species of bears. The chemical name of ursodiol is 3α,7ß-dihydroxy-5ß-cholan-24-oic (C24H40O4)
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on pack
8.3 Packaging information
10 Blister strips of 10 tablets each
8.4 Storage and handing instructions
- Store in a cool, dry & dark place.
- Keep out of reach of children.
- Tablet should be swallowed whole & not to be broken, chewed or crushed.
9.0 Patient counselling information
- Ursomax tablet should be taken after a meal with a glass of milk or water.
- Eat a healthy diet, exercise regularly, and avoid alcohol intake.
- Diarrhea may occur as a side effect. Drink plenty of fluids and inform your doctor if diarrhea persists or if you find blood in your stools.
- Your doctor may monitor your liver function and bilirubin levels every month for the next 3 months after the start of therapy, and every 6 months thereafter.
- Do not stop taking the medication without talking to your doctor.
12.0 Date of Revision
28.08.2024
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
1. What URSOMAX is and what it is used for
2. What you need to know before you take URSOMAX
3. How to take URSOMAX
4. Possible side effects
5. How to store URSOMAX
6. Contents of the pack and other information
1. WHAT URSOMAX IS AND WHAT IT IS USED FOR
The active ingredient in Ursomax tablet is Ursodeoxycholic Acid, which is a naturally occurring substance (bile acid) normally present in the bile.
Ursodeoxycholic Acid influences the composition of the bile, by which cholesterol gallstones can be solved. The effects of Ursodeoxycholic Acid in primary biliary cholangitis and cystic fibrosis can be explained by various mechanisms, such as a protective effect on the liver cells and an effect on the immune system.
1. URSOMAX Tablets is used in patients:
- with small gallstones
- who do not want to undergo surgery or are not eligible for gallstone surgery
- in whom too much cholesterol has been found in the bile
2. URSOMAX Tablets is used in patients with primary biliary cholangitis. Primary biliary cholangitis is a disease in which liver tissue is damaged by an impaired drainage of the bile.
3. URSOMAX Tablets is used in children aged 6 years to 18 years old with liver and biliary diseases caused by cystic fibrosis. Cystic fibrosis, also referred to as mucoviscidosis, is an inherited disorder in which the mucus is particularly tough in the whole body. This may cause, among other conditions, clogging and inflammation in the liver and in the bile ducts.
2. WHAT YOU NEED TO KNOW BEFORE YOU TAKE URSOMAX
Do not use URSOMAX
- You have an acute inflammation of the gallbladder or the bile ducts.
- You have a narrowing or blockage of the bile ducts.
- You suffer from often occurring cramp-like pain in the upper abdomen (biliary colic).
- You have calcified gallstones that do not transmit X-rays.
- You have a gall bladder that cannot properly constrict any more.
- You are allergic to bile acids or any of the other ingredients of this medicine.
- You have an active gastric or duodenal ulcer.
Children
In children with disrupted biliary drainage due to production of connective tissue in the bile duct (biliary atresia) in whom the bile flow is not restored by healing or by an artificial bile duct (portoenterostomy).
Warnings and precautions
This medicine must be used under medical supervision.
Your doctor should examine your liver every 4 weeks during the first three months of treatment. Then this should be done every 3 months. Apart from allowing for identification of responders and non-responders in patients being treated for primary biliary cholangitis, this monitoring would also enable an early detection of potential hepatic deterioration, particularly in patients with advanced primary biliary cholangitis.
When used for dissolving gallstones:
In order to be able to assess the therapeutic progression of the dissolution of gallstones and to timely identify a possible calcification of the stones, the gall bladder, depending on the size of the stones, should be visualized 6 to 10 months after the start of the treatment (oral cholecystography) with total image and occlusions and in the standing and lying position (ultrasound control).
If the gallbladder cannot be visualized on X-rays, or in cases of calcified gallstones, impaired contractility of the gall bladder or frequent episodes of biliary colic, the treatment with Ursodeoxycholic Tablets should be discontinued.
When used for the treatment of advanced primary biliary cholangitis:
In very rare cases decompensation of liver cirrhosis is observed which partially decreased after treatment discontinuation. Women who use URSOMAX for dissolving gallstones should stop using the birth control pill and other methods used to prevent pregnancy, because the hormones in the birth control pill can promote the production of gallstones.
When you are in the final stage of primary biliary cholangitis it can in very rare cases occur that your liver function is strongly reduced. The liver function will partly recover after stopping the treatment.
If you experience problems with diarrhoea, your doctor will reduce the dose. If the diarrhoea persists, your doctor may decide to stop the treatment.
Contact your doctor or pharmacist before you start to use this medicine.
Other medicines and URSOMAX Tablets
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines. This also applies for medicines obtained without a prescription.
The effect of the following drugs can be influenced (interactions):
A reduced effect of the following medicines is possible when using this medicine:
- Medicines binding stomach acid based on aluminium hydroxide and bile acid binding substances (colestyramine, colestipol) can bind the ursodeoxycholic acid and thereby prevent its absorption. Therefore, these medicines may not be taken simultaneously with URSOMAX, but always 2 hours before or after.
- Ursodeoxycholic acid can reduce the absorption of ciprofloxacin, dapsone (antibiotics) and nitrendipine (antihypertensive agent) from the intestine. When one of these resources has been used simultaneously with URSOMAX, your doctor will carefully supervise you.
An intensified effect of the following medicines is possible when using this medicine:
- URSOMAX may increase the absorption of cyclosporine from the intestine: if necessary, the dosing guided by the cyclosporine concentration should be adjusted in the blood.
Oestrogens, oral contraceptives ("the pill") and cholesterol-lowering agents (such as clofibrate) may promote the forming of gallstones, and can thus counteract the effect of the treatment of gallstones with URSOMAX Tablets
Inform your doctor or pharmacist, if you are taking or have recently taken other medicines. This also applies for medicines obtained without a prescription.
Pregnancy and breast-feeding
If you are pregnant or breastfeeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Pregnancy:
You should not use this medicine during pregnancy unless your doctor considers it absolutely necessary.
Women of child-bearing potential
Even if you are not pregnant, you should discuss this possibility with your doctor, because women of childbearing age may only be treated, if they use a reliable method of contraception. Non-hormonal contraception or contraception with low dose oestrogens is recommended. However, if you use this medicine for solving gallstones, you may only use non-hormonal contraception, because hormonal contraception promotes the formation of gallstones.
Breast-feeding
Tell your doctor if you are breastfeeding or about to start breastfeeding. According to few documented cases of breastfeeding women milk levels of ursodeoxycholic acid in milk are very low and probably no adverse reactions are to be expected in breastfed infants.
Driving and using machines
URSOMAX has no or negligible influence on driving ability or the ability to operate machines.
3. HOW TO USE URSOMAX
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
Your doctor will determine your dose based on your body weight.
Take the tablets after a meal with a glass of milk or a small snack. Take the prescribed number of tablets distributed throughout the day.
1. Dissolving of gallstones:
Four to six tablets of 150 mg, two to three tablets of 300 mg or two tablets of 450 mg per day (600- 900 mg ursodeoxycholic acid per day).
Two tablets should always be taken after dinner.
When taking a dose two or three times daily: for example, one tablet after lunch and two tablets after dinner.
When taking one dose of two tablets daily: take both tablets preferably two hours after dinner one hour before going to sleep.
2. Primary biliary cholangitis (damage to liver tissue by impaired bile flow):
- Phase I-III: four to eight tablets of 150 mg, two to four tablets of 300 mg, or two tablets of 450 mg per day (600-1200 mg ursodeoxycholic acid per day).
Take the prescribed dose in two to three servings per day after meals.
- Phase IV: On the basis of liver function examination your doctor will determine if a normal dosage, as in phase I-III, or the half of this dose will be prescribed to you.
Children with cystic fibrosis aged 6 to 18 years
3. Disorders of liver and biliary tract caused by cystic fibrosis (mucoviscidosis):
four to ten tablets of 150 mg, two to five tablets of 300 mg, or two to three tablets of 450 mg per day (600-1500 mg ursodeoxycholic acid per day).
The tablets with a score can be divided if you have problems in swallowing because of the size of the tablets, so that one half tablet can be taken twice directly in sequence.
If you notice that Ursodeoxycholic Acid Tablets is too strong or on the contrary too weak, consult your doctor or pharmacist.
If you take more URSOMAX than you should
You should tell your doctor if you have taken more Ursodeoxycholic Acid Tablets than you should. It is unlikely that you will notice any problems but you may experience diarrhoea.
If you forget to take URSOMAX
Take the prescribed amount at the next regular taking time. Do not take a double dose to make up for a forgotten dose.
How long should you use URSOMAX Tablets?
The duration of the treatment depends on the size of the gallstone, but is usually not shorter than three to four months. Treatment should not be interrupted prematurely; even if the symptoms have disappeared.
Only an X-ray or an ultrasound scan can show that the gallstones are completely dissolved. After it has been shown with the aid of an echogram that the gallstones have disappeared completely, the treatment should still be continued for three to four months.
The use of Ursodeoxycholic Acid in the treatment of primary biliary cholangitis and disorders of the liver and the biliary system as a result of cystic fibrosis will usually be maintained continuously, in order to continue to maintain the protective effect of Ursodeoxycholic Acid.
Do you have any further questions on the use of this medicine? Please contact your doctor or pharmacist.
4. POSSIBLE SIDE EFFECTS
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Common side effects (occurring in fewer than 1 in 10 patients but more than 1 in 100 patients)
- pasty stools or diarrhoea
Very rare side effects (may affect up to 1 in 10,000 people)
- in the treatment of primary biliary cholangitis: severe pain in the right upper abdomen, severe deterioration (decompensation) of the liver cirrhosis which partially decreases after cessation of treatment;
- calcification of gallstones;
- urticaria (hives)
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. HOW TO STORE URSOMAX
- Store in a cool, dry & dark place.
- Keep out of reach of children.
- Tablet should be swallowed whole & not to be broken, chewed or crushed.
6. CONTENTS OF THE PACK AND OTHER INFORMATION
What URSOMAX contains
Ursomax 150
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………150 mg
Excipients……………………………………………..…...q.s.
Ursomax 300
Each uncoated tablet contains
Ursodeoxycholic acid IP.……………………………300 mg
Excipients……………………………………………..…...q.s.
Ursomax 450 SR
Each prolonged-release uncoated tablet contains
Ursodeoxycholic acid IP.……………………………450 mg
Excipients……………………………………………..…...q.s.
What URSOMAX looks like and contents of the pack
10 Blister strips of 10 tablets each
Marketing authorisation holder and manufacturer
Zuventus Healthcare Ltd
Kamerey Bhasmay, Elaka Pakyong,
Rangpo, East-Sikkim 737 132.
Registered Trade Mark of Zuventus.
This leaflet was last revised in 08/2023
For More Information About This Product
Pansa DSR Capsules
1.0 Generic name
Pantoprazole Gastro-resistant and Domperidone Prolonged-release Capsules IP [40 mg + 30 mg]
2.0 Qualitative and quantitative composition
Each hard gelatin capsule contains :
Pantoprazole Sodium IP
equivalent to Pantoprazole 40 mg
(As Gastro-resistant pellets)
Colour : Indigo Carmine
Domperidone IP 30 mg
(As Prolonged-release pellets)
Colour : Lake of Sunset Yellow
Approved colours used in the capsule shell.
3.0 Pharmaceutical form and strength
Capsule, Pantoprazole (40 mg) and Domperidone (30 mg).
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment gastro-esophageal reflux disease (GERD) not responding adequately to Pantoprazole alone.
4.2 Posology and method of administration
1 Capsule to be administered once daily.
Pansa DSR capsules should be administered on empty stomach, preferably in the morning or at least 1 hour prior to meal. The capsules should be swallowed whole with water and not to be opened, chewed or crushed.
Pediatric patients
Pansa DSR Capsules are not recommended for use in children.
4.3 Contraindications
- Patients with known hypersensitivity to Pantoprazole or to any substituted Benzimidazole derivative or to Domperidone or to any component of the formulation.
- In patients receiving Rilpivirine-containing products.
- Prolactin-releasing pituitary tumor (prolactinoma).
- In patients with gastrointestinal hemorrhage, mechanical obstruction or perforation (i.e. when stimulation of the gastric motility could be harmful).
- In patients with moderate or severe hepatic impairment.
- In patients who have known existing prolongation of cardiac conduction intervals, particularly QTc.
- Patients with significant electrolyte disturbances (hypokalemia, hyperkalemia, hypomagnesemia) or underlying cardiac disease such as congestive heart failure (CHF).
- Co-administration with QT-prolonging drugs.
- Co-administration with potent CYP3A4 inhibitors.
4.4 Special warnings and precautions for use
Pantoprazole
Presence of gastric malignancy
In adults, symptomatic response to therapy with Pantoprazole does not preclude the presence of gastric malignancy. Consider additional follow-up and diagnostic testing in adult patients who have a suboptimal response or an early symptomatic relapse after completing treatment with a Pantoprazole.
Acute interstitial nephritis
Acute interstitial nephritis has been observed in patients taking Pantoprazole. Acute interstitial nephritis may occur at any point during Pantoprazole therapy and is generally attributed to an idiopathic hypersensitivity reaction. Discontinue Pantoprazole if acute interstitial nephritis develops.
Clostridium difficile-associated diarrhea (CDAD)
Published observational studies suggest that Pantoprazole therapy may be associated with an increased risk of CDAD, especially in hospitalized patients. This diagnosis should be considered for diarrhea that does not improve.
Risk of bone fractures
Proton pump inhibitors (PPIs), especially if used in high doses and over long durations (> 1 year), may modestly increase the risk of hip, wrist and spine fracture, predominantly in the elderly or in presence of other recognized risk factors. Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated. Patients at risk of osteoporosis should receive care according to current clinical guidelines and they should have an adequate intake of Vitamin D and Calcium.
Cutaneous and systemic lupus erythematosus
Cutaneous lupus erythematosus (CLE) and systemic lupus erythematosus (SLE) have been reported in patients taking PPIs, including Pantoprazole. These events have occurred as both new onset and an exacerbation of existing autoimmune disease. The most common form of CLE reported in patients treated with PPIs was subacute CLE (SCLE) and occurred within weeks to years after continuous drug therapy in patients ranging from infants to the elderly. The occurrence of CLE with previous PPI treatment may increase the risk of CLE with other PPIs. Systemic lupus erythematosus (SLE) is less commonly reported than CLE in patients receiving PPIs. The majority of patients presented with rash. If lesions occur, especially in sun-exposed areas of the skin, and if accompanied by arthralgia, the patient should seek medical help promptly and Pantoprazole therapy should be stopped immediately. Most patients improve with discontinuation of the PPI alone in 4 to 12 weeks.
Cyanocobalamin (Vitamin B12) deficiency
Generally, daily treatment with any acid-suppressing medication over a long period of time (e.g. longer than 3 years) may lead to malabsorption of Vitamin B12 caused by hypo- or achlorhydria. Rare reports of Cyanocobalamin deficiency occurring with acidsuppressing therapy have been reported in the literature. This diagnosis should be considered if clinical symptoms consistent with Cyanocobalamin deficiency are observed.
Hypomagnesemia
Hypomagnesemia, symptomatic and asymptomatic, has been reported rarely in patients treated with PPIs for at least 3 months, in most cases after a year of therapy. Serious adverse events include tetany, arrhythmias, and seizures. In most patients, treatment of hypomagnesemia required magnesium replacement and discontinuation of the PPI. For patients expected to be on prolonged treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g. diuretics), monitoring of magnesium levels prior to initiation of PPI treatment and periodically thereafter should be considered.
Domperidone
Cardiovascular effects
Domperidone has been associated with prolongation of the QT interval on the electrocardiogram. During post-marketing surveillance, there have been very rare cases of QT prolongation and torsades de pointes in patients taking Domperidone. These reports included patients with confounding risk factors, electrolyte abnormalities and concomitant treatment which may have been contributing factors.
Epidemiological studies showed that Domperidone was associated with an increased risk of serious ventricular arrhythmias or sudden cardiac death. A higher risk was observed in patients older than 60 years, patients taking daily doses greater than 30 mg and patients concurrently taking QT-prolongation drugs or CYP3A4 inhibitors.
Domperidone is contraindicated in patients with known existing prolongation of cardiac conduction intervals, particularly QTc, in patients with significant electrolyte disturbances (hypokalaemia, hyperkalaemia, hypomagnesaemia), or bradycardia, or in patients with underlying cardiac diseases such as congestive heart failure (CHF) due to increased risk of ventricular arrhythmia. Electrolyte disturbances or bradycardia are known to be conditions increasing the proarrythmic risk. Treatment with Domperidone should be stopped if signs or symptoms occur that may be associated with cardiac arrhythmia, and the patients should consult their physician. Patients should be advised to promptly report any cardiac symptoms.
Use with Apomorphine
Domperidone is contraindicated with QT prolonging drugs including Apomorphine, unless the benefit of the co-administration with Apomorphine outweighs the risks.
Use in infants and children
Although neurological side effects are rare, the risk of neurological side effects are higher in young children since metabolic
functions and the blood-brain barrier are not fully developed in the first months of life. Overdosing may cause extrapyramidal
symptoms in children, but other causes should be taken into consideration.
4.5 Interaction with other medicinal products and other forms of interaction
Pantoprazole
Antiretroviral drugs
The effect of PPIs on antiretroviral drugs is variable. The clinical importance and the mechanisms behind these interactions are not always known.
- Decreased exposure of some antiretroviral drugs (e.g. Rilpivirine, Atazanavir and Nelfinavir) when used concomitantly with Pantoprazole may reduce antiviral effect and promote the development of drug resistance. Concomitant use of Rilpivirine containing products with Pantoprazole is contraindicated. Also, concomitant use of nelfinavir with Pantoprazole should be avoided.
- Increased exposure of other antiretroviral drugs (e.g. Saquinavir) when used concomitantly with Pantoprazole may increase toxicity
- There are other antiretroviral drugs which do not result in clinically relevant interactions with Pantoprazole.
Coumarin anticoagulants / Warfarin
There has been post-marketing reports of increased international normalized ratio (INR) and prothrombin time in patients receiving PPIs, including Pantoprazole and Warfarin concomitantly. Increases in INR and prothrombin time may lead to abnormal bleeding and even death. Monitor INR and prothrombin time and adjust the dose of Warfarin if needed, to maintain the target INR range.
Clopidogrel
Concomitant administration of Pantoprazole and Clopidogrel in healthy subjects had no clinically important effect on exposure to the active metabolite of Clopidogrel or Clopidogrel-induced platelet inhibition. No dose adjustment of Clopidogrel is necessary when administered with an approved dose of Pantoprazole.
Methotrexate
Literature suggests that concomitant use of PPIs with Methotrexate (primarily at high dose) may elevate and prolong serum levels of Methotrexate and/or its metabolite, possibly leading to Methotrexate toxicities. A temporary withdrawal of Pantoprazole therapy may be considered in some patients receiving high-dose of Methotrexate.
Drugs for which gastric pH can affect bioavailability (iron salts, Erlotinib, Dasatinib, Nilotinib, Mycophenolate Mofetil, and Ketoconazole)
Pantoprazole causes long-lasting inhibition of gastric acid secretion. Therefore, Pantoprazole may reduce absorption of other drugs where gastric pH is an important determinant of their bioavailability.
Mycophenolate Mofetil (MMF)
Co-administration of Pantoprazole Sodium in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, Mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving Pantoprazole therapy and MMF. Use Pantoprazole with caution in transplant patients receiving MMF.
Drug / laboratory tests interactions
False positive urine tests for THC
There have been reports of false positive urine screening tests for Tetrahydrocannabinol (THC) in patients receiving PPIs, including Pantoprazole. An alternative confirmatory method should be considered to verify positive results.
Increased Chromogranin A (CgA) level
Increase in CgA may interfere with investigations for neuroendocrine tumours. To avoid this interference, Pantoprazole treatment should be stopped for at least 5 days before CgA measurements. If CgA and gastrin levels have not returned to reference range after initial measurement, measurements should be repeated 14 days after cessation of Pantoprazole treatment.
Domperidone
The main metabolic pathway of Domperidone is through CYP3A4. In vitro data suggest that the concomitant use of drugs that significantly inhibit this enzyme may result in increased plasma levels of Domperidone. There is increased risk of occurrence of QT-interval prolongation, due to pharmacodynamic and/or pharmacokinetic interactions.
Concomitant use of the following drugs is contraindicated.
A. QTc-prolonging medicinal products
- Anti-arrhythmic class IA (e.g. Disopyramide, Hydroquinidine, Quinidine).
- Anti-arrhythmic class III (e.g. Amiodarone, Dofetilide, Dronedarone, Ibutilide, Sotalol).
- Certain antipsychotics (e.g. Haloperidol, Pimozide, Sertindole). • Certain antidepressants (e.g. Citalopram, Escitalopram).
- Certain antibiotics (e.g. Erythromycin, Levofloxacin, Moxifloxacin, Spiramycin).
- Certain antifungal agents (e.g. Pentamidine).
- Certain antimalarial agents (e.g. Halofantrine, Lumefantrine).
- Certain gastrointestinal medicines (e.g. Cisapride, Dolasetron, Prucalopride).
- Certain antihistaminic (e.g. Mequitazine, Mizolastine).
- Certain medicines used in cancer (e.g. Toremifene, Vandetanib, Vincamine).
- Other medicines (e.g. Bepridil, Diphemanil, Methadone).
B. Potent CYP3A4 inhibitors (regardless of their QT prolonging effects)
- Protease inhibitors.
- Systemic azole antifungals.
- Some macrolides (e.g. Erythromycin, Clarithromycin and Telithromycin).
Concomitant use of the following drugs is not recommended.
- Moderate CYP3A4 inhibitors (e.g. Diltiazem, Verapamil and some macrolides).
Concomitant use of the following drugs requires caution.
- Caution with bradycardia and hypokalaemia-inducing drugs, as well as with the following macrolides involved in QT-interval prolongation : Azithromycin and Roxithromycin.
Ketoconazole / Erythromycin and QTc prolongation
Separate in vivo pharmacokinetic / pharmacodynamic interaction studies with oral Ketoconazole or oral erythromycin in healthy subjects confirmed a marked inhibition of Domperidone's CYP3A4 mediated first pass metabolism by these drugs (as both of these drugs significantly inhibit CYP3A4 enzyme). Both the Cmax and AUC of Domperidone at steady state were increased approximately three-fold in each of these interaction studies. In these studies, concomitant use of Domperidone and Ketoconazole or Erythromycin resulted in increase in QTc, over the observation period.
4.6 Use in special populations
Pregnancy
Pansa DSR capsule should be used during pregnancy only if the potential benefit justifies the possible risk to the fetus.
Breast-feeding
Pansa DSR capsules should not be used during breast feeding. Accordingly, a decision should be made whether to discontinue nursing or to discontinue / abstain from therapy, taking into account the benefit of the drug to the mother.
4.7 Effects on ability to drive and use machines
Both, Pantoprazole and Domperidone have no or negligible influence on the ability to drive and use machines. However, adverse reactions such as dizziness and visual disturbances may occasionally occur in patients on PPIs drug therapy. If affected, patients should not drive or use machines.
4.8 Undesirable effects
Pantoprazole
Clinical trials experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug may not reflect the rates observed in clinical practice. Common adverse reactions reported with Pantoprazole therapy in clinical trials with frequency > 2% include : Headache, diarrhoea, nausea, abdominal pain, vomiting, flatulence, dizziness, and arthralgia. Additional adverse reactions reported in clinical trials with a frequency of ≤ 2% include : Body as a whole :
Allergic reaction, pyrexia, photosensitivity reaction, facial edema
Gastrointestinal : Constipation, dry mouth, hepatitis
Hematologic : Leukopenia, thrombocytopenia
Metabolic / nutritional : Elevated creatine kinase (CK), generalized edema, elevated triglycerides, elevated liver enzymes
Musculoskeletal : Myalgia
Nervous : Depression, vertigo
Skin and appendages : Urticaria, rash, pruritus
Special senses : Blurred vision
Post-marketing experience
Acute kidney injury as an adverse drug reaction reported with the use of proton pump inhibitors. The following adverse reactions have been identified during post-approval use of Pantoprazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
General disorders and administration conditions : Asthenia, fatigue, malaise
Hematologic : Pancytopenia, agranulocytosis
Hepatobiliary disorders : Hepatocellular damage leading to jaundice and hepatic failure
Immune system disorders : Anaphylaxis (including anaphylactic shock), SLE
Infections and infestations : Clostridium difficile-associated diarrhea
Investigations : Weight changes
Metabolism and nutritional disorders : Hyponatremia, hypomagnesemia
Musculoskeletal disorders : Rhabdomyolysis, bone fracture
Nervous system : Ageusia, dysgeusia
Psychiatric disorders : Hallucination, confusion, insomnia, somnolence
Renal and urinary disorders : Interstitial nephritis
Skin and subcutaneous tissue disorders : Severe dermatologic reactions, including erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis (TEN), angioedema (Quincke’s edema) and CLE
Domperidone
Central nervous system : As the pituitary gland is outside the blood-brain barrier, Domperidone may cause an increase in prolactin levels. In rare cases this hyperprolactinaemia may lead to neuro-endocrinological side effects such as galactorrhoea, gynaecomastia and amenorrhoea.
Extrapyramidal side effects are very rare in neonates and infants, and exceptional in adults. These side effects reverse spontaneously and completely as soon as the treatment is stopped. Other central nervous system-related effects of convulsion, agitation and somnolence also are very rare and primarily reported in infants and children.
General disorders
Uncommon : Asthenia.
Immune system disorder
Not known : Anaphylactic reactions including anaphylactic shock and angioedema
Psychiatric disorders
Uncommon : Anxiety, loss of libido;
Not known : Agitation, nervousness
Nervous system disorders
Uncommon : Somnolence, headache;
Not known : Extrapyramidal disorder, convulsions
Eye disorders Not known : Oculogyric crisis Cardiac disorders
Not known : Ventricular arrhythmias, QTc prolongation, Torsade de Pointes, sudden cardiac death. Gastrointestinal disorders
Common : Dry mouth;
Uncommon : Diarrhea Skin and subcutaneous tissue disorders
Uncommon : Rash, pruritus;
Not known : Urticaria, angioedema Reproductive system and breast disorders
Uncommon : Breast pain, breast tenderness, galactorrhoea;
Not known : Gynaecomastia, amenorrhoea
Renal and urinary disorders
Not known : Urinary retention Investigations
Not known : Abnormal liver function test, increased blood prolactin
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com
Website : https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Symptoms
Symptoms of overdose are agitation, altered consciousness, convulsions, disorientation, somnolence, and extrapyramidal reactions.
Treatment
There is no specific antidote, but in the event of overdose, gastric lavage as well as the administration of activated charcoal, may be useful. Close medical supervision and supportive therapy is recommended. Anticholinergic, Antiparkinson drugs may be helpful in controlling the extrapyramidal reactions.
5.0 Pharmacological properties
5.1 Mechanism of action
Pantoprazole
Pantoprazole is a proton pump inhibitor (PPI) class of anti-secretory agent. Pantoprazole is a lipophilic weak base that crosses the parietal cell membrane and enters the acidic parietal cell canaliculus where it becomes protonated, producing the active metabolite sulfenamide. Sulfenamide forms an irreversible covalent bond with two sites of the H+/K+-ATPase enzyme located on the gastric parietal cell. Thus, Pantoprazole suppress the final step in gastric acid (Hydrochloric Acid - HCl) production by covalently binding to the H+/K+-ATPase enzyme (also called as proton pump) system at the secretory surface of the gastric parietal cell. This effect leads to inhibition of both basal and stimulated gastric acid secretion, irrespective of the stimulus. The binding to the H+/K+-ATPase results in duration of anti-secretory effect that persists longer than 24 hours.
Domperidone
Domperidone is a dopamine receptor (D2) antagonist. Domperidone act predominantly on peripheral dopamine receptors and produces anti-emetic and gastro-kinetic effects. Domperidone does not readily cross the blood-brain barrier (BBB). Thus, in Domperidone users, especially in adults, extrapyramidal side effects are very rare (unlike metoclopramide). Anti-emetic effect of Domperidone is due to a combination of peripheral (gastro-kinetic) effects and antagonism of dopamine receptors in the chemoreceptor trigger zone (CTZ), which lies outside the BBB in the area postrema. Oral Domperidone also increases lower esophageal sphincter (LES) pressure, thus, improve antroduodenal motility and accelerate gastric emptying. There is no effect on gastric secretion.
5.2 Pharmacodynamic properties
With a single oral dose of 20 to 80 mg of Pantoprazole, a dose-dependent decrease in gastric acid secretion occurs. Following the initial oral dose of 40 mg Pantoprazole, a 51% mean inhibition was achieved by 2.5 hours. With once-a-day dosing for 7 days, the mean inhibition was increased to 85%. Acid secretion had returned to normal within a week after the last dose of Pantoprazole; there was no evidence of rebound hypersecretion.
Pantoprazole reduces acidity in the stomach and thereby increases gastrin in proportion to the reduction in acidity. The increase in gastrin is reversible. Since Pantoprazole binds to the enzyme distal to the receptor level, it can inhibit hydrochloric acid secretion independently of stimulation by other substances (Acetylcholine, histamine and gastrin). The fasting gastrin values increase under Pantoprazole. On short-term use, in most cases they do not exceed the upper limit of normal. During long-term treatment, gastrin levels double in most cases.
Domperidone
Prokinetic effect : The prokinetic (gastro-kinetic) properties of Domperidone are related to its peripheral dopamine receptor blocking action.
Antiemetic effect : Domperidone produces antiemetic effect by blocking dopamine receptors (D2) peripherally. Inhibition of peripheral D2 receptor signaling prevents or relieves various GI symptoms, such as nausea and vomiting, and also relieves reflux and other symptoms associated with upper GI disorders.
5.3 Pharmacokinetic properties
Pantoprazole
Absorption
Like other PPIs, Pantoprazole is an acid-labile drug and therefore, administered orally in the form of gastro-resistant pellets. Absorption of Pantoprazole, therefore, begins in the intestine only after the pellets leave the stomach. After administration of a single or multiple oral doses of Pantoprazole 40 mg, the peak plasma concentration of Pantoprazole was achieved in approximately 2.5 hours, and Cmax was 2.5 mcg/ml. Peak serum concentration (Cmax) and area under the serum concentration time curve (AUC) increases in a dose-dependent manner (with dose range from 10 to 80 mg). Pantoprazole does not accumulate, and its pharmacokinetics are unaltered with multiple daily dosing. Pantoprazole undergoes little first-pass metabolism, resulting in an absolute bioavailability of approximately 77%.
Effect of antacid / food
Pantoprazole absorption is not affected by concomitant administration of antacids. Administration of Pantoprazole with food may delay its absorption up to 2 hours or longer; however, the Cmax and the extent of Pantoprazole absorption (AUC) are not altered. Thus, Pantoprazole may be taken without regard to timing of meals.
Distribution
The apparent volume of distribution of Pantoprazole is approximately 11 to 23.6 liters, distributing mainly in extracellular fluid.
The plasma protein binding of Pantoprazole is about 98%, primarily to albumin.
Metabolism
Pantoprazole is extensively metabolized in the liver through the cytochrome P450 (CYP) system. The main metabolic pathway is demethylation, by CYP2C19, with subsequent sulfation; other metabolic pathways include oxidation by CYP3A4. There is no evidence that any of the Pantoprazole metabolites have significant pharmacologic activity.
Excretion
Renal elimination represents the major route of excretion (about 80 %) for the metabolites of Pantoprazole, the rest is excreted with the faeces. There is no renal excretion of unchanged Pantoprazole. The main metabolite in both the serum and urine is desmethyl Pantoprazole which is conjugated with Sulphate. Following oral administration, the serum concentration of Pantoprazole declines biexponentially, with a terminal elimination half-life of approximately one hour.
Domperidone
Pharmacokinetics of Domperidone in sustained release formulation is not available. Conventional formulation of Domperidone (i.e. immediate release) has following pharmacokinetic properties :
Absorption
Domperidone is rapidly absorbed after oral administration, with peak plasma concentrations occurring at approximately 1 hour after dosing. The Cmax and AUC values of Domperidone increased proportionally with dose in the 10 mg to 20 mg dose range. The low absolute bioavailability of oral Domperidone (approximately 15%) is due to an extensive first-pass metabolism in the gut and liver.
Distribution
Oral Domperidone does not appear to accumulate or induce its own metabolism. The peak plasma concentration (Cmax) of 18 ng/ml to 21 ng/ml occurs 1.5 hours (Tmax) after the oral dose. Domperidone is 91 to 93% bound to plasma proteins. Distribution studies with Domperidone have shown wide tissue distribution, but low brain concentration.
Metabolism
Domperidone undergoes rapid and extensive hepatic metabolism by hydroxylation and N-dealkylation. In vitro metabolism experiments with diagnostic inhibitors revealed that CYP3A4 is a major form of cytochrome P-450 involved in the N-dealkylation of Domperidone, whereas CYP3A4, CYP1A2 and CYP2E1 are involved in Domperidone aromatic hydroxylation.
Excretion
After oral dose, Domperidone is excreted mainly by renal (31%) and biliary (66%) routes. The proportion of the drug excreted unchanged is small (10% of fecal excretion and approximately 1% of urinary excretion). The plasma half-life after a single oral dose is 7 to 9 hours in healthy subjects, but is prolonged in patients with severe renal insufficiency.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Pantoprazole
Carcinogenesis
In a 24-month carcinogenicity study, Sprague-Dawley rats were treated orally with Pantoprazole doses of 0.5 to 200 mg/kg/day, about 0.1 to 40 times the exposure on a body surface area basis of a 50 kg person dosed with 40 mg/day. In the gastric fundus, treatment with 0.5 to 200 mg/kg/day produced enterochromaffin-like (ECL) cell hyperplasia and benign and malignant neuroendocrine cell tumors in a dose-related manner. In the forestomach, treatment with 50 and 200 mg/kg/day (about 10 and 40 times the recommended human dose on a body surface area basis) produced benign squamous cell papillomas and malignant squamous cell carcinomas. Rare gastrointestinal tumors associated with Pantoprazole treatment included an adenocarcinoma of the duodenum with 50 mg/kg/day and benign polyps and adenocarcinomas of the gastric fundus with 200 mg/kg/day. In the liver, treatment with 0.5 to 200 mg/kg/day produced dose-related increases in the incidences of hepatocellular adenomas and carcinomas. In the thyroid gland, treatment with 200 mg/kg/day produced increased incidences of follicular cell adenomas and carcinomas for both male and female rats. In a 24-month carcinogenicity study, B6C3F1 mice were treated orally with doses of 5 to 150 mg/kg/day of Pantoprazole, 0.5 to 15 times the recommended human dose based on body surface area. In the liver, treatment with 150 mg/kg/day produced increased incidences of hepatocellular adenomas and carcinomas in female mice. Treatment with 5 to 150 mg/kg/day also produced gastric-fundic ECL cell hyperplasia. A 26-week p53 +/-transgenic mouse carcinogenicity study was not positive.
Domperidone
Safety margins in in vitro proarrhythmic models (isolated Langendorff perfused heart) exceeded the free plasma concentrations in humans at maximum daily dose (10 mg administered 3 times a day) by 9- up to 45-fold. In in vivo models the no effect levels for QTc prolongation in dogs and induction of arrhythmias in a rabbit model sensitized for torsade de 13 pointes exceeded the free plasma concentrations in humans at maximum daily dose (10 mg administered 3 times a day) by more than 22-fold and 435-fold, respectively. In the anesthetized guinea pig model following slow intravenous infusions, there were no effects on QTc at total plasma concentrations of 45.4 ng/ml, which are 3-fold higher than the total plasma levels in humans at maximum daily dose (10 mg administered 3 times a day). At a high, maternally toxic dose (more than 40 times the recommended human dose), teratogenic effects were seen in the rat. No teratogenicity was observed in mice and rabbits. Development abnormalities observed in rats at a high exposure. Risk of carcinogenicity, mutagenicity or sensitisation cannot be excluded.
7.0 Description
Each capsule of Pansa DSR contains 40 mg of Pantoprazole (in a gastro-resistant form) and 30 mg of Domperidone (in a prolonged release form) for oral administration in adults.
Pantoprazole
Pantoprazole Sodium is the Sodium salt form of Pantoprazole. Pantoprazole is a substituted Benzimidazole, proton pump inhibitor (PPI) class of anti-secretory agents which suppressesgastric acid production.
Molecular weight : 405.40 g/mol
Molecular formula : C16H14F2N3NaO4S
Chemical name : Sodium; 5-(difluoromethoxy)-2-[(3,4-dimethoxypyridin-2- yl) methylsulfinyl] benzimidazol-1-ide Domperidone Domperidone is a dopamine receptor antagonist drug with antiemetic and gastrokinetic properties. Domperidone is white or almost white powder which is slightly soluble in water. Molecular weight : 425.90 g/mol Molecular formula : C22H24ClN5O2 Chemical name : 6-chloro-3-[1-[3-(2-oxo-3H-benzimidazol-1-yl)propyl]piperidin-4-yl]-1H-benzimidazol-2-one.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Alu-Alu blister strip of 10 capsules.
8.4 Storage and handling instructions
Store protected from moisture at a temperature not exceeding 25°C.
Keep out of reach of children.
9.0 Patient counseling information
- Instruct patients to take Pansa DSR Capsule exactly as prescribed by doctor. Do not change the dose or stop therapy without consulting to your doctor.
- Instruct patients to swallow Pansa DSR Capsule as a whole with water and not to open, chew or crush the capsules.
- If you miss a dose, take it as soon as possible. If it is almost time for your next dose, do not take the missed dose. Take the next dose at your regular time. Do not take 2 capsules per doses at the same time to make up for the missed dose.
- Pregnant women can use this medicine only if essential and in consultation with their doctor.
- Advise nursing mothers to avoid use of this medicine during lactation or not to breastfeed their infants while on drug therapy.
- This medicine is not recommended for use in children.
- Instruct patients not to share this medication with other people even though symptoms are similar. It may harm them.
- Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Pansa DSR Capsules and certain other medicines can interact with each other causing serious side effects.
12.0 Date of issue
04 July 2024
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
1.What PANSA DSR is and what it is used for
2. What you need to know before you take PANSA DSR
3. How to take PANSA DSR
4. Possible side effects
5. How to store PANSA DSR
6. Contents of the pack and other information
1. What PANSA DSR is and what it is used for
Pansa DSR Capsule is a combination of two medicines: Domperidone and Pantoprazole. Domperidone is a prokinetic which works on the upper digestive tract to increase the movement of the stomach and intestines, allowing the food to move more easily through the stomach. Pantoprazole is a proton pump inhibitor (PPI) which works by reducing the amount of acid in the stomach which helps in the relief of acid-related indigestion and heartburn
PANSA DSR is used for the treatment of Gastroesophageal reflux disease (occurs when stomach acid repeatedly flows back into the tube connecting your mouth and stomach that is esophagus)
2. What you need to know before you take PANSA DSR
Do not take PANSA DSR if you are
- If you are allergic to pantoprazole, domperidone or to any of the other ingredients listed in the formulation
- If you are allergic to medicines containing other proton pump inhibitors.
- You have black, tarry bowel motions (stools) or notice blood in your bowel motions. This could be a sign of bleeding in the stomach or intestines
- You have a blockage or tear in your intestines
- You have a tumour of the pituitary gland called a prolactinoma.
- have a disorder known as phenylketonuria (a metabolic disorder) orodispersible tablets should not be used as they contain aspartamine
- if you have a moderate or severe liver disease
- if your ECG (electrocardiogram) shows a heart problem called "prolonged QT corrected interval"
- if you have or had a problem where your heart cannot pump the blood round your body as well as it should (condition called heart failure).
- if you have a problem that gives you a low level of potassium or magnesium, or a high level of potassium in your blood.
Do not take PANSA DSR tablets if any of the above applies to you. If you are not sure, talk to your doctor or pharmacist before taking this medicine. If you are not sure if any of the above apply to you, talk to your doctor or pharmacist before taking PANSA DSR tablet. Do this even if they have applied in the past.
Warnings and precautions
Talk to your doctor, pharmacist or nurse before taking PANSA DSR
- If you have severe liver problems. Please tell your doctor if you ever had problems with your liver in the past. He will check your liver enzymes more frequently, especially when you are taking Pantoprazole as a long-term treatment. In the case of a rise of liver enzymes the treatment should be stopped.
- If you have reduced body stores or risk factors for reduced vitamin B12 and receive long-term treatment with pantoprazole. As with all acid reducing agents, pantoprazole may lead to a reduced absorption of vitamin B12.
- If you are taking HIV protease inhibitors such as atazanavir (for the treatment of HIV-infection) at the same time as pantoprazole, ask your doctor for specific advice.
- Taking a proton pump inhibitor like pantoprazole, especially over a period of more than one year, may slightly increase your risk of fracture in the hip, wrist or spine. Tell your doctor if you have osteoporosis or if you are taking corticosteroids (which can increase the risk of osteoporosis).
- If you are on Pantoprazole for more than three months it is possible that the levels of magnesium in your blood may fall. Low levels of magnesium can be seen as fatigue, involuntary muscle contractions, disorientation, convulsions, dizziness, increased heart rate. If you get any of these symptoms, please tell your doctor promptly. Low levels of magnesium can also lead to a reduction in potassium or calcium levels in the blood. Your doctor may decide to perform regular blood tests to monitor your levels of magnesium.
- If you have ever had a skin reaction after treatment with a medicine similar to Pantoprazole that reduces stomach acid.
- If you get a rash on your skin, especially in areas exposed to the sun tell your doctor as soon as you can, as you may need to stop your treatment with Pantoprazole. Remember to also mention any other ill-effects like pain in your joints.
- if you are due to have a specific blood test (Chromogranin A).
- if you suffer from liver problems (liver function impairment or failure)
- if you suffer from kidney problems (kidney function impairment or failure).
It is advisable to ask your doctor for advice in case of prolonged treatment as you may need to take a lower dose or take this medicine less often, and your doctor may want to examine you regularly. Domperidone may be associated with an increased risk of heart rhythm disorder and cardiac arrest. This risk may be more likely in those over 60 years old or taking doses higher than 30mg per day. The risk also increases when domperidone is given together with some drugs. Tell your doctor or pharmacist if you are taking drugs to treat infection (fungal infections or bacterial infection) and/or if you have heart problems or AIDS/HIV. Domperidone should be used at the lowest effective dose. While taking domperidone, contact your doctor if you experience heart rhythm disorders such as palpitations, trouble breathing, loss of consciousness. Treatment with domperidone should be stopped.
Adolescents weighing less than 35 kg and children
PANSA DSR should not be given to adolescents 12 years of age and older weighing less than 35 kg, or in any children less than 12 years of age, as it is not effective in these age groups.
Tell your doctor immediately, before or after taking this medicine, if you notice any of the following
- An unintentional loss of weight
- Vomiting, particularly if repeated
- Vomiting blood; this may appear as dark coffee grounds in your vomit
- You notice blood in your stools; which may be black or tarry in appearance
- Difficulty in swallowing or pain when swallowing
- You look pale and feel weak (anaemia)
- Chest pain, Stomach pain, severe and/or persistent diarrhoea, because this medicine
- has been associated with a small increase in infectious diarrhoea.
Your doctor may decide that you need some tests to rule out malignant disease because pantoprazole also alleviates the symptoms of cancer and could cause delay in diagnosing it. If your symptoms continue in spite of your treatment, further investigations will be considered. If you take Pantoprazole on a long-term basis (longer than 1 year) your doctor will probably keep you under regular surveillance. You should report any new and exceptional symptoms and circumstances whenever you see your doctor.
Other medicines and PANSA DSR
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines, including medicines obtained without a prescription. This is because Pantoprazole may influence the effectiveness of other medicines, so tell your doctor if you are taking:
- Medicines such as ketoconazole, itraconazole and posaconazole (used to treat fungal infections) or erlotinib (used for certain types of cancer) because Pantoprazole may stop these and other medicines from working properly.
- Warfarin and phenprocoumon, which affect the thickening, or thinning of the blood. You may need further checks.
- Medicines used to treat HIV-infection, such as atazanavir.
- Methotrexate (used to treat rheumatoid arthritis, psoriasis, and cancer) – if you are taking methotrexate your doctor may temporarily stop your Pantoprazole treatment because pantoprazole can increase levels of methotrexate in the blood.
- Fluvoxamine (used to treat depression and other psychiatric diseases) – if you are taking fluvoxamine your doctor may reduce the dose.
- Rifampicin (used to treat infections).
- St John’s wort (Hypericum perforatum) (used to treat mild depression).
Do not take Domperidone tablet if you are taking medicine to treat:
- fungal infections such as azole antifungals, specifically oral ketoconazole, fluconazole or voriconazole
- bacterial infections, specifically erythromycin, clarithromycin, telithromycin,moxifloxacin, pentamidine (these are antibiotics)
- heart problems or high blood pressure (e.g., amiodarone, dronedarone, quinidine, disopyramide, dofetilide, sotalol, diltiazem, verapamil)
- psychoses (e.g., haloperidol, pimozide, sertindole)
- depression (e.g., citalopram, escitalopram)
- gastro-intestinal disorders (e.g., cisapride, dolasetron, prucalopride)
- allergy (e.g., mequitazine, mizolastine)
- malaria (in particular halofantrine)
- AIDS/HIV (protease inhibitors)
- Hepatitis C (e.g., telaprevir)
- cancer (e.g., toremifene, vandetanib, vincamine)
- certain other medicines (e.g., bepridil, diphemanil, methadone)
Before you use Domperidone tablet and apomorphine, your doctor will ensure that you tolerate both medicines when used simultaneously. Ask your doctor or specialist for a personalised advice. Tell your doctor or pharmacist if you are taking drugs to treat infection, heart problems or AIDS/HIV. It is important to ask your doctor or pharmacist if Domperidone tablet is safe for you when you are taking any other medicines, including medicines obtained without prescription.
Pregnancy
Pantoprazole: There are no adequate and well-controlled studies in pregnant women.
Domperidone: There are limited post-marketing data of its use in pregnant women. A study in rats has shown reproductive toxicity at a high, maternally toxic dose.
Because animal reproduction studies are not always predictive of human response, PANSA DSR tablet should be used during pregnancy only if clearly needed.
Lactation
There is insufficient data on the excretion of pantoprazole in human milk but excretion into human milk has been reported.
Domperidone is excreted in human milk and breast-fed infants receive less than 0.1% of the maternal weight-adjusted dose. Caution should be exercised in case of QTc prolongation risk factor in breast-fed infants.
A decision should be made whether to discontinue breast-feeding or to discontinue/abstain from PANSA DSR therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the women.
Driving and using machines
PANSA DSR has no or negligible influence on the ability to drive and use machines. If you experience side effects like dizziness or disturbed vision, you should not drive or operate machines.
3. How to take PANSA DSR
One capsule daily. The capsule should not be chewed or crushed, and should be swallowed whole 1 hour before a meal with some water.
Pediatric
PANSA DSR tablets are not recommended in children below 12 years of age.
Hepatic impairment
No dosage adjustment is needed in patients with mild to severe hepatic impairment. In patients with severe liver impairment, the liver enzymes should be monitored regularly during treatment with pantoprazole, particularly on long-term use.
In the case of a rise of the liver enzymes, the treatment should be discontinued. Doses higher than 40 mg/day have not been studied in patients with hepatic impairment
Renal impairment
In subjects with renal insufficiency (creatinine clearance<30 ml/min/1.73m2) the elimination half-life of domperidone was increased from 7.4 to 20.8 hours. Since very little unchanged drug (approx. 1%) is excreted via the kidneys, it is unlikely that the dose of a single administration needs to be adjusted in patients with renal insufficiency. However, on repeated administration, the dosing frequency of PANSA DSR should be reduced to once or twice daily depending on severity of the impairment.
If you take more PANSA DSR than you should
Consult your doctor or pharmacist. There are no known symptoms of overdose.
If you forget to take PANSA DSR
Do not take a double dose to make up for a forgotten dose. Take your next, normal dose at the usual time.
If you stop taking PANSA DSR
Do not stop taking these tablets without first talking to your doctor or pharmacist.
4. Possible Side Effects
Pantoprazole
Like all medicines, this medicine can cause side effects, although not everybody gets them.
If you get any of the following side effects, stop taking these tablets and tell your doctor immediately, or contact the casualty department at your nearest hospital:
Serious allergic reactions (frequency rare: may affect up to 1 in 1,000 people): swelling of the tongue and/or throat, difficulty in swallowing, hives (nettle rash), difficulties in breathing, allergic facial swelling (Quincke’s oedema / angioedema), severe dizziness with very fast heartbeat and heavy sweating.
Serious skin conditions (frequency not known: frequency cannot be estimated from the available data): blistering of the skin and rapid deterioration of your general condition, erosion (including slight bleeding) of eyes, nose, mouth/lips or genitals (Stevens-Johnson-Syndrome, Lyell-Syndrome, Erythema multiforme), and sensitivity to light.
Other serious conditions (frequency not known: frequency cannot be estimated from the available data): yellowing of the skin or whites of the eyes (severe damage to liver cells, jaundice) or fever, rash, and enlarged kidneys sometimes with painful urination, and lower back pain (serious inflammation of the kidneys), possibly leading to kidney failure.
Other side effects are:
Common (may affect up to 1 in 10 people) Benign polyps in the stomach.
Uncommon (may affect up to 1 in 100 people) Headache; dizziness; diarrhoea; feeling sick, vomiting; bloating and flatulence (wind); constipation; dry mouth; abdominal pain and discomfort; skin rash, exanthema, eruption; itching; feeling weak, exhausted or generally unwell; sleep disorders; fracture in the hip, wrist or spine.
Rare (may affect up to 1 in 1,000 people) Distortion or complete lack of the sense of taste; disturbances in vision such as blurred vision; hives; pain in the joints; muscle pains; weight changes; raised body temperature; high fever; swelling of the extremities (peripheral oedema); allergic reactions; depression; breast enlargement in males.
Very Rare (may affect up to 1 in 10,000 people) Disorientation.
Not known (frequency cannot be estimated from the available data) Hallucination, confusion (especially in patients with a history of these symptoms); decreased sodium level in blood, decreased magnesium level in blood, feeling of tingling, prickling, pins and needles, burning sensation or numbness, rash, possibly with pain in the joints, inflammation in the large bowel, that causes persistent watery diarrhoea.
Side effects identified through blood tests:
Uncommon (may affect up to 1 in 100 people) an increase in liver enzymes.
Rare (may affect up to 1 in 1,000 people) an increase in bilirubin; increased fat levels in blood; sharp drop in circulating granular white blood cells, associated with high fever.
Very Rare (may affect up to 1 in 10,000 people) a reduction in the number of blood platelets, which may cause you to bleed or bruise more than normal; a reduction in the number of white blood cells, which may lead to more frequent infections; coexisting abnormal reduction in the number of red and white blood cells, as well as platelets.
Domperidone
Uncommon (may affect up to 1 in 100 people): Involuntary movements of the face or arms and legs, excessive trembling, excessive muscle stiffness or muscle spasm
Not known (frequency cannot be estimated from the available data):
- Seizures
- a type of reaction that may occur soon after administration and is recognised by skin rash, itching, shortness of breath, and/or a swollen face.
- a severe hypersensitivity reaction that may occur soon after administration that is characterised by hives, itching, flushing, fainting, and difficulty breathing among other possible symptoms.
- disorders of the cardiovascular system: heart rhythm disorders (rapid or irregular heart beat) have been reported; if this happens, you should stop the treatment immediately.
Domperidone may be associated with an increased risk of heart rhythm disorder and cardiac arrest. This risk may be more likely in those over 60 years old or taking doses higher than 30 mg per day. Domperidone should be used at the lowest effective dose. Stop treatment with Domperidone and contact your doctor immediately if you experience any of the unwanted events described above.
Other unwanted effects that have been observed with Domperidone are listed below:
Common (may affect up to 1 in 10 people): Dry mouth
Uncommon (may affect up to 1 in 100 people): anxiety, agitation, nervousness, loss of interest in sex or diminished interest in sex, headache, sleepiness, diarrhoea, rash, itchiness, hives, Painful or tender breasts, milk discharge from breasts, a general feeling of weakness, feeling dizzy
Not known (frequency cannot be estimated from the available data): Upward movement of the eyes, stopped menstrual periods in women, enlarged breasts in men, inability to urinate, changes in certain laboratory test results, restless legs syndrome (uncomfortable feeling, with an irresistible urge to move your legs, and sometimes arms and other parts of your body).
Some patients who have used Domperidone for conditions and dosages requiring medical oversight have experienced the following unwanted effects: Restlessness; swollen or enlarged breasts, unusual discharge from breasts, irregular menstrual periods in women, difficulty breastfeeding, depression, hypersensitivity.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to store PANSA DSR
Store protected from light & moisture at a temperature not exceeding 30°C.
Keep out of reach of children.
Do not use the tablets after the expiry date is stated on the label. Medicines should not be disposed of via wastewater or household waste.
Ask your pharmacist how to dispose of medicines no longer required. These measures will help to protect the environment.
6. Contents of the pack and other information
What Zosa Junior gastro-resistant granules for oral suspension contains
Each hard gelatin capsule contains:
Pantoprazole Sodium IP
equivalent to Pantoprazole- 40 mg
(As Gastro-resistant pellets)
Colours : Lake of Sunset Yellow
Domperidone IP -30 mg
(As Prolonged-release pellets)
Colour : Indigo Carmine
Excipients q.s.
Approved colours used in the capsule shell.
Packaging
A blister strip of 10 capsules