Plecasoft Tablets


1.0 Generic name
Plecanatide Tablets 3 mg
2.0 Qualitative and quantitative composition
Each uncoated tablet contains :
Plecanatide 3 mg
3.0 Dosage form & strength
Tablet 3 mg
4.0 Clinical particulars
4.1 Therapeutic indications
Indicated in adults for the treatment of
- Chronic idiopathic constipation (CIC)
- Irritable bowel syndrome with constipation (IBS-C)
4.2 Posology and method of administration
The recommended dosage of Plecasoft for the treatment of CIC and IBS-C is 3 mg taken orally once daily. Preparation and administration instructions
- Plecasoft can be taken with or without food.
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take two doses at the same time.
- Swallow a tablet whole for each dose.
- For adult patients with swallowing difficulties, Plecasoft tablets can be crushed and administered orally either in applesauce or with water or administered with water via a nasogastric or gastric feeding tube. Mixing Plecasoft crushed tablets in other soft foods or in other liquids has not been tested.
Oral administration in applesauce
- In a clean container, crush the Plecasoft tablet to a powder and mix with 1 teaspoonful of room temperature applesauce.
- Consume the entire tablet-applesauce mixture immediately. Do not store the mixture for later use.
Oral administration in water
- Place the Plecasoft tablet in a clean cup.
- Pour approximately 30 mL of room temperature water into the cup.
- Mix by gently swirling the tablet and water mixture for at least 10 seconds. The Plecasoft tablet will fall apart in the water.
- Swallow the entire contents of the tablet water mixture immediately.
- If any portion of the tablet is left in the cup, add another 30 mL of water to the cup, swirl for at least 10 seconds, and swallow immediately.
- Do not store the tablet-water mixture for later use.
Administration with water via a nasogastric or gastric feeding tube
- Place the Plecasoft tablet in a clean cup with 30 mL of room temperature water.
- Mix by gently swirling the tablet and water mixture for at least 15 seconds. The Plecasoft tablet will fall apart in the water.
- Flush the nasogastric or gastric feeding tube with 30 mL of water using a catheter tip syringe.
- Draw up the mixture using the syringe and immediately administer via the nasogastric or gastric feeding tube. Do not reserve for future use.
- If any portion of the tablet is left in the cup, add another 30 mL of water to the cup, swirl for at least 15 seconds, and using the same syringe, administer via the nasogastric or gastric feeding tube.
- Using the same or a fresh syringe, flush the nasogastric or gastric feeding tube with at least 10 mL of water.
4.3 Contraindications
- Patients less than 6 years of age due to the risk of serious dehydration.
- Patients with known or suspected mechanical gastrointestinal obstruction.
4.4 Special warnings and precautions for use
Risk of serious dehydration in pediatric patients
Plecanatide is contraindicated in patients less than 6 years of age. The safety and effectiveness of Plecanatide in patients less than 18 years of age have not been established Due to increased intestinal expression of GC-C, patients less than 6 years of age may be more likely than patients 6 years of age and older to develop severe diarrhea and its potentially serious consequences. Avoid the use of Plecanatide in patients 6 years to less than 18 years of age. Diarrhea If severe diarrhea occurs, suspend dosing and rehydrate the patient.
4.5 Drugs interactions
Neither plecanatide nor its active metabolite inhibited the cytochrome P450 (CYP) enzymes 2C9 and 3A4, and they did not induce CYP3A4 in vitro. Plecanatide and its active metabolite were neither substrates nor inhibitors of the transporters Pglycoprotein (P-gp) or breast cancer resistance protein (BCRP) in vitro.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Pregnancy
Plecanatide and its active metabolite are negligibly absorbed systemically following oral administration and maternal use is not expected to result in fetal exposure to the drug. The available data on Plecanatide use in pregnant women are not sufficient to inform any drug associated risks for major birth defects and miscarriage. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes.
Lactation
There is no information regarding the presence of plecanatide in human milk, or its effects on milk production or the breastfed infant. Plecanatide and its active metabolite are negligibly absorbed systemically following oral administration. It is unknown whether the negligible systemic absorption of plecanatide by adults will result in a clinically relevant exposure to breastfed infants. Exposure to plecanatide in breastfed infants has the potential for serious adverse effects. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Plecanatide and any potential adverse effects on the breastfed infant from Plecanatide or from the underlying maternal condition.
Pediatric use
Plecanatide is contraindicated in pediatric patients less than 6 years of age. Avoid use of Plecanatide in patients 6 years to less than 18 years of age. The safety and effectiveness of Plecanatide in patients less than 18 years of age have not been established. Because of increased intestinal expression of GC-C, patients less than 6 years of age may be more likely than patients 6 years of age and older to develop diarrhea and its potentially serious consequences. Plecanatide is contraindicated in patients less than 6 years of age.
Geriatric use
The safety and effectiveness of Plecanatide in patients greater than 65 years of age have not been established.
4.7 Effects on ability to drive and use machines
Plecanatide has no or negligible influence on the ability to drive and use machines. During treatment with Plecanatide, dizziness has been reported as less common adverse reaction. Therefore, patients who experience dizziness should be cautious while driving or using machines.
4.8 Undesirable effects

Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.co.in/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is no specific treatment to the event of overdose. In the event of overdose, the patient should be treated symptomatically and supportive measures instituted as required.
5.0 Pharmacological properties
5.1 Mechanism of action
Plecanatide is a structural analog of human uroguanylin, and similarly to uroguanylin, plecanatide functions as a guanylate cyclase-C (GC-C) agonist. Both plecanatide and its active metabolite bind to GC-C and act locally on the luminal surface of the intestinal epithelium. Activation of GC-C results in an increase in both intracellular and extracellular concentrations of cyclic guanosine monophosphate (cGMP). Elevation of extracellular cGMP has been associated with a decrease in the activity of pain-sensing nerves in animal models of visceral pain. Elevation of intracellular cGMP stimulates secretion of chloride and bicarbonate into the intestinal lumen, mainly through activation of the cystic brosis transmembrane conductance regulator (CFTR) ion channel, resulting in increased intestinal fluid and accelerated transit.
5.2 Pharmacodynamic properties
Food effect
Subjects who received either a low-fat, low calorie (LF-LC) meal or a high fat, high calorie (HF-HC) meal reported looser stools than fasted subjects up to 24 hours after a single dose of plecanatide 9 mg (3 times the recommended dose). In clinical studies, plecanatide was administered with or without food.
5.3 Pharmacokinetic properties
Absorption
Plecanatide was minimally absorbed with negligible systemic availability following oral administration. Concentrations of plecanatide and its active metabolite in plasma were below the limit of quantitation in the majority of analyzed plasma samples after an oral plecanatide dose of 3 mg. Therefore, standard pharmacokinetic parameters such as AUC, Cmax, and half-life (t1/2) could not be calculated.
Distribution
Given that plecanatide concentrations following clinically relevant oral doses were not measurable, plecanatide is expected to be minimally distributed in tissues. Oral plecanatide was localized to the GI tract where it exerted its effects as a GC-C agonist with negligible systemic exposure. Plecanatide exhibited little to no binding to human serum albumin or human α-1-acid glycoprotein.
Metabolism
Plecanatide was metabolized in the GI tract to an active metabolite by loss of the terminal leucine moiety. Both plecanatide and the metabolite were proteolytically degraded within the intestinal lumen to smaller peptides and naturally occurring amino acids.
Excretion
Plecanatide and its active metabolite were not measurable in plasma following administration of the recommended clinical doses.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
The carcinogenic potential of plecanatide was assessed in 2-year carcinogenicity studies in mice and rats. Plecanatide was not tumorigenic in mice at oral doses up to 90 mg/kg/day or in rats at oral doses up to 100 mg/kg/day. Limited systemic exposure to plecanatide was achieved at the tested dose levels in animals, whereas no detectable exposure occurred in humans. Therefore, animal and human doses should not be compared directly for evaluating relative exposure. Plecanatide was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, in vitro mouse lymphoma mutation assay, or the in vivo mouse bone marrow micronucleus assay. Plecanatide had no effect on fertility or reproductive function in male or female mice at oral doses of up to 600 mg/kg/day.
7.0 Description
Plecasoft (plecanatide) is a guanylate cyclase-C (GC-C) agonist. Plecanatide is a 16 amino acid peptide with the following chemical name : L-Leucine, L-asparaginyl-L-α-aspartyl-L-α-glutamyl-L-cysteinyl-L-α-glutamyl-L-leucyl-L-cysteinyl-L-valyl-L-asparaginyl-L-valyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-, cyclic (4 12), (7 15)-bis (disufide).The molecular formula of plecanatide is C65H104N18O26S4 and the molecular weight is 1682 Daltons.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Alu-Alu blister strip of 10 tablets.
8.4 Storage and handling instructions
Store at a temperature below 25°C.
Keep out of reach of children.
9.0 Patient counselling information
Advise patients :
Diarrhea
To stop Plecasoft and contact their healthcare provider if they experience severe diarrhea.
Accidental ingestion
Accidental ingestion of Plecasoft in children, especially in children less than 6 years of age, may result in severe diarrhea and dehydration. Instruct patients to take steps to store Plecasoft securely and out of reach of children and to dispose of unused Plecanatide.
Administration and handling instructions
- To take Plecasoft once daily with or without food
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take two doses at the same time.
- To swallow Plecasoft tablets whole.
- If adult patients have swallowing difficulties, Plecasoft tablets can be crushed and administered orally in either applesauce or with water, or administered with water via a nasogastric or gastric feeding tube. • To keep Plecasoft in a dry place. Protect from moisture. For bottles, keep Plecasoft in the original bottle. Do not remove desiccant from the bottle. Do not subdivide or repackage. Remove and discard polyester coil after opening. Keep bottles closed tightly.
12.0 Date of issue
21 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Plecasoft is and what it is used for
- What you need to know before you take Plecasoft
- How to take Plecasoft
- Possible side effects
- How to store Plecasoft
- Contents of the pack and other information
1. What Plecasoft is and what it is used for
Plecasoft contains the active ingredient plecanatide, which works directly in the intestine to help increase fluid and support regular bowel movements. It acts like a natural substance in your body to draw water into the intestines, making stools softer and easier to pass. This makes stools softer and easier to pass. It helps relieve symptoms like hard stools, bloating, and stomach discomfort. Plecasoft is used in adults to treat long-term(chronic) constipation and constipation caused by irritable bowel syndrome (IBS-C).
2. What you need to know before you take Plecasoft
Do not take Plecasoft if you:
- Are less than 6 years of age (due to risk of serious dehydration)
- Have a known or suspected mechanical obstruction in your gut/intestine
Warnings and precautions
Talk to your doctor before taking Plecasoft if:
- You are between 6 to 18 years of age, as it is not recommended in this age group.
- You experience severe diarrhoea, in which case treatment should be stopped and you should consult your doctor.
Children and adolescents
Do not give this medicine to children under 6 years of age. Avoid use in children and adolescents aged 6 to less than 18 years.
Other medicines and Plecasoft
Plecasoft has a low likelihood of interacting with other medicines. However, always inform your doctor or pharmacist if you are taking or have recently taken any other medicines.
Pregnancy and breast-feeding
- This medicine is not absorbed significantly in the body; however, its safety in pregnancy is not fully established.
- It is unknown whether Plecanatide passes into breast milk. Speak to your doctor before using this medicine if you are pregnant or breastfeeding.
Driving and using machines
Plecasoft has no or negligible influence on the ability to drive or use machines. However, dizziness has been reported in some cases.
3. How to take Plecasoft
Dosage
The recommended dose is one tablet (3 mg) once daily, with or without food.
Administration
- Swallow the tablet whole.
- If you have trouble swallowing:
- You may crush the tablet and mix with 1 teaspoon of applesauce or 30 mL of water.
- It can also be administered via a nasogastric or gastric feeding tube.
- Always consume or administer the mixture immediately. Do not store it for later use.
- Follow your doctor’s instructions.
If you miss a dose
Skip the missed dose. Take your next dose at the regular time. Do not take two doses at the same time.
4. Possible side effects
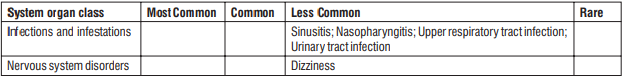
Less common:
- Nausea
- Abdominal distension
- Flatulence
- Sinusitis
- Nasopharyngitis
- Upper respiratory tract infection
- Urinary tract infection
- Dizziness
- Increased liver enzyme levels
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Plecasoft
- Store below 25°C.
- Keep in the original packaging to protect from moisture.
- Keep out of sight and reach of children.
- Do not use after the expiry date stated on the pack.
6. Contents of the pack and other information
What Plecasoft contains
- The active substance is plecanatide 3 mg per tablet.
What Plecasoft looks like and contents of the pack
- Plecasoft is available in Alu-Alu blister packs of 10 tablets.
Marketing Authorisation Holder
Zuventus Healthcare Ltd. Plot Y2, CTS No. 358/A2, Near Nahur Railway Station, Nahur (West), Mumbai - 400 078, India.
This leaflet was last revised in May 2025.
For More Information About This Product
Floresp - OX Nasal Spray
1.0 Generic name
Fluticasone Furoate & Oxymetazoline Hydrochloride Nasal Spray
2.0 Qualitative and quantitative composition
Each spray delivers :
Fluticasone Furoate 27.5 mcg
Oxymetazoline Hydrochloride IP 50 mcg
Composition :
Fluticasone Furoate 0.028% w/w
Oxymetazoline Hydrochloride IP 0.050% w/w
Benzalkonium Chloride Solution IP 0.030% w/w
3.0 Dosage form & strength
Nasal Spray, Fluticasone Furoate 27.5 mcg & Oxymetazoline Hydrochloride 50 mcg
4.0 Clinical particulars
4.1 Therapeutic indication
For the treatment of symptoms of allergic rhinitis with nasal congestion.
4.2 Posology and method of administration
The recommended dose is two spray actuations (27.5 micrograms of. Fluticasone Furoate and 50 mcg of Oxymetazoline Hydrochloride per spray actuation) in each nostril once daily (total daily dose, 110 micrograms of Fluticasone Furoate and 200 mcg of Oxymetazoline Hydrochloride). Once adequate control of symptoms is achieved, dose reduction to one spray actuation in each nostril (total daily dose 55 micrograms of Fluticasone Furoate and 100 mcg of Oxymetazoline Hydrochloride) may be effective for maintenance.
Fluticasone Furoate and Oxymetazoline Hydrochloride Nasal Spray is for administration by the intranasal route only.
The intranasal device should be shaken before use. The device is primed by pressing the mist release button for at least six spray actuations (until a fine mist is seen), whilst holding the device upright. Re-priming (approximately 6 sprays until a fine mist is seen) is only necessary if the cap is left off for 5 days or the intranasal device has not been used for 30 days or more.
Children < 12 years of age
Fluticasone Furoate and Oxymetazoline Hydrochloride Nasal Spray should not be used in children less than 12 years of age.
Elderly (≥ 65 years old)
No dose adjustment is required in this population.
Renal impairment
No dose adjustment is required in this population.
Hepatic impairment
No dose adjustment is required in this population.
4.3 Contraindications
- Hypersensitivity to Fluticasone Furoate, Oxymetazoline or any other ingredients of this medicine.
- Age less than 12 years.
- In patients who have undergone recent trans-nasal surgery.
- In patients with chronic nasal inammation with very dry nasal passages (rhinitis sicca or atrophic rhinitis).
- In patients with cardiovascular disease, hyperthyroidism, angle closure glaucoma or prostatic enlargement.
4.4 Special warnings and precautions for use
Keep the spray away from the eyes.
Systemic corticosteroid effects
Systemic effects of nasal corticosteroid may occur, particularly at high doses prescribed for prolonged periods. These effects are much less likely tooccur than with oral corticosteroids and may vary in individual patients and between different corticosteroid preparations. Potential systemic effects may include Cushing's syndrome, Cushingoid features, adrenal suppression, growth retardation in children and adolescents, cataract, glaucoma and more rarely, a range of psychological or behavioural effects including psychomotor hyperactivity, sleep disorders, anxiety, depression or aggression (particularly in children). Treatment with higher than recommended doses of nasal corticosteroids may result in clinically significant adrenal suppression. If there is evidence for higher than recommended doses being used, then additional systemic corticosteroid cover should be considered during periods of stress or elective surgery. Fluticasone furoate 110 micrograms once daily has not been associated with hypothalamic-pituitary-adrenal (HPA) axis suppression in adult or adolescent subjects. However, the dose of intranasal fluticasone furoate should be reduced to the lowest dose at which effective control of the symptoms of rhinitis is maintained. As with all intranasal corticosteroids, the total systemic burden of corticosteroids should be considered whenever other forms of corticosteroid treatment are prescribed concurrently. If there is any reason to believe that adrenal function is impaired, care must be taken when transferring patients from systemic steroid treatment to fluticasone furoate.
Visual disturbance
Visual disturbance may be reported with systemic and topical corticosteroid use. If a patient present with symptoms such as blurred vision or other visual disturbances, the patient should be considered for referral to an ophthalmologist for evaluation of possible causes which may include cataract, glaucoma or rare diseases such as central serous chorioretinopathy (CSCR) which have been reported after use of systemic and topical corticosteroids.
Growth retardation
Growth retardation has been reported in children receiving nasal corticosteroids at licensed doses. A reduction in growth velocity has been observed in children treated with fluticasone furoate 110 micrograms daily for one year. Therefore, children should be maintained on the lowest possible efficacious dose which delivers adequate symptom control. It is recommended that the growth of children receiving prolonged treatment with nasal corticosteroids is regularly monitored. If growth is slowed, therapy should be reviewed with the aim of reducing the dose of nasal corticosteroid if possible, to the lowest dose at which effective control of symptoms is maintained.
Patients on ritonavir
Concomitant administration with ritonavir is not recommended because of the risk of increased systemic exposure of fluticasone Furoate.
4.5 Drug interactions
Interaction with CYP3A inhibitors
Fluticasone furoate is rapidly cleared by extensive first-pass metabolism mediated by the cytochrome P450 3A4.
Based on data with another glucocorticoid (fluticasone propionate), that is metabolised by CYP3A4, coadministration with ritonavir is not recommended because of the risk of increased systemic exposure of fluticasone furoate. Caution is recommended when co-administering fluticasone furoate with potent CYP3A inhibitors including cobicistat containing products as an increase in the risk of systemic side effects is expected. Co-administration should be avoided unless the benefit outweighs the increased risk of systemic corticosteroid side effects, in which case patients should be monitored for systemic corticosteroid side effects. In a drug interaction study of intranasal fluticasone furoate with the potent CYP3A4 inhibitor ketoconazole there were more subjects with measurable fluticasone furoate concentrations in the ketoconazole group (6 of the 20 subjects) compared to placebo (1 out of 20 subjects). This small increase in exposure did not result in a statistically significant difference in 24-hour serum cortisol levels between the two groups.
The enzyme induction and inhibition data suggest that there is no theoretical basis for anticipating metabolic interactions between fluticasone furoate and the cytochrome P450 mediated metabolism of other compounds at clinically relevant intranasal doses. Therefore, no clinical studies have been conducted to investigate interactions of fluticasone furoate on other drugs Monoamine oxidase inhibitors (MAOIs), nonselective beta adrenergic antagonists, or tricyclic antidepressants.
Use of Oxymetazoline containing nasal spray in combination with monoamine oxidase inhibitors (MAOIs), nonselective beta adrenergic antagonists, or tricyclic antidepressants may cause hypertension and is not recommended.
4.6 Use in special populations
Pregnancy
There are no adequate data from the use of fluticasone furoate and Oxymetazoline containing nasal sprays in pregnant women. In animal studies glucocorticoids have been shown to induce malformations including cleft palate and intra-uterine growth retardation. This is not likely to be relevant for humans given recommended nasal doses which results in minimal systemic exposure.
Breast-feeding
It is unknown whether nasal administered fluticasone furoate or Oxymetazoline is excreted in human breast milk. Administration of Fluticasone Furoate and Oxymetazoline Hydrochloride Nasal Spray to women who are breast-feeding should only be considered if the expected benet to the mother is greater than any possible risk to the child.
Fertility
There are no fertility data in humans.
4.7 Effects on ability to drive and use machines
Fluticasone Furoate and Oxymetazoline Hydrochloride Nasal Spray has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
The most common AEs reported with the use of Fluticasone Furoate and Oxymetazoline containing Nasal Spray are epistaxis, nasal ulceration, nasal dryness & discomfort, sneezing and headache. The other AEs include : rhinalgia, transient ocular changes, vision blurred, nasal septum perforation, dry mouth, stomatitis, dry throat, local irritation, insomnia, sedation, anxiety, irritability, nausea, hypertension, irregular heart rate, increased heart rate, restlessness, somnolence, fatigue, hypersensitivity reactions including anaphylaxis, angioedema, rash, and urticaria. Prolonged use may cause rebound congestion and rhinitis medicamentosa.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.co.in/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Overdosage may give rise to local irritation and rebound congestion. Treatment need only be symptomatic
5.0 Pharmacological properties
5.1 Pharmacodynamic properties
Fluticasone furoate is a synthetic tri-fluorinated corticosteroid that possesses a very high affinity for the glucocorticoid receptor and has a potent anti-inflammatory action.
Oxymetazoline is a direct-acting sympathomimetic amine. It acts on alpha-adrenergic receptors in the vessels of the nasal mucosa producing vasoconstriction and decongestion.
5.2 Pharmacokinetic properties
Fluticasone Furoate
Absorption: Fluticasone furoate undergoes incomplete absorption and extensive first-pass metabolism in the liver and gut resulting in negligible systemic exposure. The intranasal dosing of 110 micrograms once daily does not typically result in measurable plasma concentrations (<10 pgl ml). The absolute bioavailability for intranasal uticasone furoate is 0.50 %, such that less than 1 microgram of fluticasone furoate would be systemically available after administration of 110 micrograms
Distribution: The plasma protein binding of fluticasone furoate is greater than 99%. Fluticasone furoate is widely distributed with volume of distribution at steady-state of, on average, 608 L.
Biotransformation: Fluticasone furoate is rapidly cleared (total plasma clearance of 58.7 I/h) from systemic circulation principally by hepatic metabolism to an inactive 17p-carboxylic metabolite (GW694301X), by the cytOchrome P450 enzyme CYP3A4. The principal route of metabolism was hydrolysis of the S-fluoromethyl carbothioate function to form the 1713- carboxylic acid metabolite. In vivo studies have revealed no evidence of cleavage of the furoate moiety to form fluticasone.
Elimination: Elimination is primarily via the faecal route following oral and intravenous administration indicative of excretion of fluticasone furoate and its metabolites via the bile. Following intravenous administration, the elimination phase half-life averaged 15.1 hours. Urinary excretion accounted for approximately 1 % and 2 % of the orally and intravenously administered dose, respectively.
Oxymetazoline Hydrochloride
Oxymetazoline enters tissues rapidly and local vasoconstriction is normally achieved within 5-10 minutes of intranasal administration. The full effect lasts for 5-6 hours and then gradually subsides over the next 6 hours. Plasma half-life is 5-8 days with 30% of any absorbed drug being excreted in the urine unchanged and 10% being excreted in the faeces
6.0 Nonclinical properties
6.1 Animal Toxicology or pharmacology
Fluticasone Furoate
Findings in general toxicology studies were similar to those observed with other glucocorticoids and are associated with exaggerated pharmacological activity. These findings are not likely to be relevant for humans given recommended nasal doses which results in minimal systemic exposure. No genotoxic effects of fluticasone furoate have been observed in conventional genotoxicity tests. Further, there were no treatment-related increases in the incidence of tumours in two-year inhalation studies in rats and mice.
Oxymetazoline Hydrochloride
There are no preclinical data of relevance
7.0 Description
Fluticasone Furoate and Oxymetazoline Hydrochloride Nasal Spray is white to off white suspension in a pre-filled intranasal sprayer. The pH of suspension is 5.0 ± 1.0 The product contains two active ingredients : Fluticasone Furoate and Oxymetazoline Hydrochloride.
The chemical name of fluticasone Furoate is (6a,1113,16a,17a)-6,9- diuoro-17-{[(uoro-methypthioicarbony1}-11-hydroxy-16-methyl-3- oxoandrosta-1,4-dien-17-y1 2-furancarboxylate. The molecular weight is 538.6.
The chemical formula of Oxymetazoline Hydrochloride is 3-[(4,5- dihydro1H-imidazol-2-yOmethyl]-6-(1,1 ,-dimethylethyl)-2,4- dimethylphenol mono-hydrochloride. Its molecular weight is 296.8 for the hydrochloride salt and 260.4 for the free base.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
FLORESP-OX sales pack contains 7 gm (70 metered doses).
8.4 Storage and handling instructions
Store in a dry & dark place at a temperature not exceeding 25°C.
Keep out of reach of children.
9.0 Patient Counselling Information
- Keep the spray away from the eyes.
- Prolonged use of spray may lead to systemic corticosteroid effects. Consult your physician for duration of use of the product.
- Do not use the product in children less than 12 years of age
- Do not use the product if you have cardiovascular disease, hyperthyroidism, angle closure glaucoma or prostatic enlargement Do not use the product if you have undergone recent trans-nasal surgery, or in case of chronic nasal inflammation with very dry nasal passages (rhinitis sicca or atrophic rhinitis).
12.0 Date of issue
30 September 2024
About Leaflet
Read all of this leaflet carefully before you start using this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others.
- If you get any side effects, talk to your doctor or pharmacist.
What is in this leaflet
- What FLORESP OX is and what it is used for
- What you need to know before you use FLORESP OX
- How to use FLORESP OX
- Possible side effects
- How to store FLORESP OX
- Contents of the pack and other information
1. What FLORESP OX is and what it is used for
FLORESP OX is a nasal spray that contains two active substances: Fluticasone Furoate: a corticosteroid that reduces inflammation. Oxymetazoline Hydrochloride: a decongestant that relieves nasal congestion.
It is used to treat symptoms of allergic rhinitis, such as
- Nasal congestion
- Sneezing
- Runny or itchy nose
2. What you need to know before you use FLORESP OX
Do not use FLORESP OX if you:
- Are allergic to Fluticasone Furoate, Oxymetazoline, or any other ingredients.
- Are under 12 years of age.
- Have had recent nasal surgery.
- Have very dry nasal passages (rhinitis sicca or atrophic rhinitis).
- Have heart disease, high blood pressure, thyroid problems, glaucoma, or prostate issues.
Warnings and precautions
Talk to your doctor before using this spray if you:
- Are taking other corticosteroids.
- Have eye problems like blurred vision or glaucoma.
- Are pregnant or breastfeeding.
- Are taking ritonavir or other strong CYP3A4 inhibitors.
Children
Do not use in children under 12 years. Long-term use may affect growth.
3. How to take Floresp OX Nasal Spray
For Adults and Teenagers (12 years and older):
- Start with 2 sprays in each nostril once a day (total of 110 micrograms).
- Once your symptoms are under control, you can reduce to 1 spray in each nostril once a day (total of 55 micrograms).
- Always use the lowest dose that works for you.
For Children (2 to 11 years old):
- Start with 1 spray in each nostril once a day (total of 55 micrograms).
- If symptoms don’t improve, your doctor may recommend 2 sprays in each nostril once a day (total of 110 micrograms).
- Once symptoms are better, go back to 1 spray in each nostril once a day.
For Elderly People:
- No special dose changes are needed.
For People with Kidney or Liver Problems:
- No dose changes are needed.
Method of administration:
- Remove the protective cap.
- Insert the nozzle into the nostril and press to spray.
- Allow excess solution to trickle down and clear your nose after 30 seconds to 1 minute.
- Repeat if necessary.
- Wash the nozzle with soapy water, wipe dry, and replace the cap. • For infants and children, use under adult supervision.
Other Precautions
- Shake the bottle before using the medicine.
- Clean your nose thoroughly before using the medicine.
- Insert the bottle tip into one nostril and close the other nostril.
- Direct the spray towards the sides of your nostril, away from the cartilage dividing the two sides of your nose.
- Breathe out gently through your mouth and repeat the same process for the other nostril.
- Avoid deep breathing as it will cause medication to go back to the throat and make it less effective.
- Avoid deep breathing as it will cause the medication to go back to the throat and make it less effective.
- Do not share the bottle with anyone else so that you do not spread germs.
If you use more Floresp OX Nasal Spray than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Floresp OX Nasal Spray
If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Floresp OX Nasal Spray
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Common side effects:
- Nosebleeds
- Nasal dryness or discomfort
- Sneezing
- Headache
Uncommon or rare side effects:
- Blurred vision
- Dry mouth or throat
- Sleep problems, anxiety, or irritability
- Allergic reactions (rash, swelling, difficulty breathing)
- Rebound congestion with prolonged use
If you experience serious side effects, stop using the spray and seek medical help immediately.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Floresp OX Nasal Spray
- Store in a dry & dark place at a temperature not exceeding 25°C.
- Keep out of reach of children.
6. Contents of the pack and other information
What FLORESP OX contains:
Fluticasone Furoate 27.5 mcg
Oxymetazoline Hydrochloride 50 mcg
Other ingredients: Benzalkonium Chloride, water, etc.
Pack size: 7 g (70 metered doses)
Revised on 06/25
For More Information About This Product
Supexa- OD 60 Tablets
1.0 Generic name
Edoxaban Tablets
2.0 Qualitative and quantitative composition
SUPEXA OD 15
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….15 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 30
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….30 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 60
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….60 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
3.0 Dosage form and strength
Film coated tablet, 15 mg / 30 mg / 60 mg
4.0 Clinical particulars
4.1Therapeutic indications
- For prevention of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation (NVAF) with one or more risk factors, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischaemic attack (TIA).
- For the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the prevention of recurrent DVT and PE in adults.
4.2 Posology and method of administration
Posology
Prevention of stroke and systemic embolism
- The recommended dose is 60 mg edoxaban once daily. Therapy with edoxaban in NVAF patients should be continued long term.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate or severe renal impairment (creatinine clearance (CrCl) 15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of the following P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- For elderly patients aged 80 years and above: dose reduced to 15 mg once daily.
Treatment of DVT, PE and prevention of recurrent DVT and PE (venous thromboembolism (VTE))
- The recommended dose is 60 mg edoxaban once daily following initial use of parenteral anticoagulant for at least 5 days. Edoxaban and initial parenteral anticoagulant should not be administered simultaneously. The duration of therapy for treatment of DVT and PE (VTE), and prevention of recurrent VTE should be individualized after careful assessment of the treatment benefit against the risk for bleeding. Short duration of therapy (at least 3 months) should be based on transient risk factors (e.g. recent surgery, trauma, immobilization) and longer durations should be based on permanent risk factors or idiopathic DVT or PE.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate to severe renal impairment (CrCl -15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Prevention of venous thromboembolism (VTE) in patients undergoing orthopedic surgery for the lower limbs - Total knee or hip replacement, hip fracture surgery:
- The usual adult dosage is 30 mg of edoxaban administered orally once daily.
- Consider reducing the dose to 15 mg once daily if
- Renal impairment (CrCl 30 mL/min and ≤ 50 mL/min).
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Edoxaban should be administered 12 hours after surgery and after confirming that there is no bleeding from the surgical wound, etc.
- Edoxaban should be at least 2 hours after the epidural catheter is removed or lumbar puncture is performed.
Table 1: Summary of Edoxaban Posology in NVAF and VTE (DVT and PE)

Missed dose
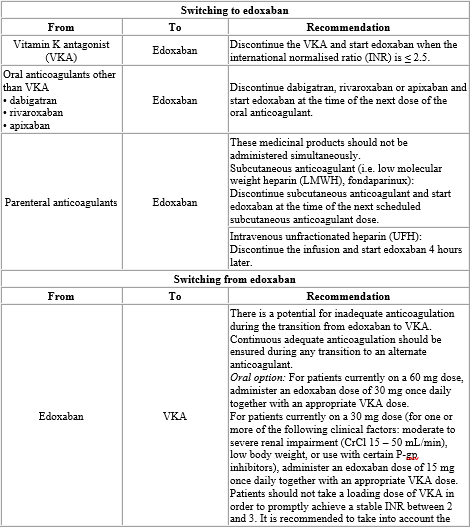
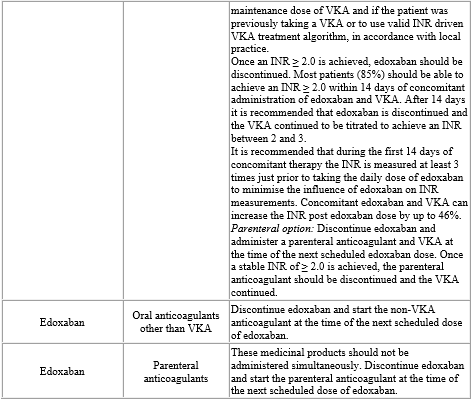
If a dose of edoxaban is missed, the dose should be taken immediately and then be continued the following day with the once-daily intake as recommended. The patient should not take double the prescribed dose on the same day to make up for a missed dose. Switching to and from edoxaban Continued anticoagulant therapy is important in patients with NVAF and VTE. There may be situations that warrant a change in anticoagulation therapy (Table 2).
Table 2: Switching of anticoagulant treatment in NVAF and VTE (DVT and PE)
Special populations
Elderly population
No dose reduction is required.
Renal impairment
Renal function should be assessed in all patients by calculating the CrCl prior to initiation of treatment with edoxaban to exclude patients with end stage renal disease (i.e. CrCl < 15 mL/min), to use the correct edoxaban dose in patients with CrCl 15 – 50 mL/min (30 mg once daily), in patients with CrCl > 50 mL/min (60 mg once daily) and when deciding on the use of edoxaban in patients with increased CrCl.
Renal function should also be assessed when a change in renal function is suspected during treatment (e.g. hypovolaemia, dehydration, and in case of concomitant use of certain medicinal products).
The method used to estimate renal function (CrCl in mL/min) during the clinical development of edoxaban was the Cockcroft-Gault method. The formula is as follows:
For creatinine in µ mol/L:
1.23× (140-age [years])× weight [kg] (× 0.85 if Female)
Serum creatinine [µmol/L]
For creatinine in mg/dL:
(140-age [years]) × weight [kg] (× 0.85 if Female)
72×Serum creatinine [mg/dL]
This method is recommended when assessing patients' CrCl prior to and during edoxaban treatment.
In patients with mild renal impairment (CrCl > 50 – 80 mL/min), the recommended dose is 60 mg edoxaban once daily.
In patients with moderate or severe renal impairment (CrCl 15 – 50 mL/min), the recommended dose is 30 mg edoxaban once daily.
In patients with end stage renal disease (ESRD) (CrCl < 15 mL/min) or on dialysis, the use of edoxaban is not recommended.
Hepatic impairment
Edoxaban is contraindicated in patients with hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
In patients with severe hepatic impairment edoxaban is not recommended.
In patients with mild to moderate hepatic impairment the recommended dose is 60 mg edoxaban once daily. Edoxaban should be used with caution in patients with mild to moderate hepatic impairment.
Patients with elevated liver enzymes (alanine aminotransferase (ALT) or aspartate transaminase (AST) > 2 x upper limit of normal (ULN)) or total bilirubin ≥ 1.5 x ULN, were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed.
Body weight
For patients with body weight ≤ 60 kg, the recommended dose is 30 mg edoxaban once daily).
Gender
No dose reduction is required.
Concomitant use of edoxaban with P-glycoprotein (P-gp) inhibitors
In patients concomitantly taking edoxaban and the following P-gp inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem, the recommended dose is 30 mg edoxaban once daily.
No dose reduction is required for concomitant use of amiodarone, quinidine or verapamil. The use of edoxaban with other P-gp inhibitors including HIV protease inhibitors has not been studied.
Patients undergoing cardioversion
Edoxaban can be initiated or continued in patients who may require cardioversion. For transoesophageal echocardiogram (TEE) guided cardioversion in patients not previously treated with anticoagulants, edoxaban treatment should be started at least 2 hours before cardioversion to ensure adequate anticoagulation. Cardioversion should be performed no later than 12 hours after the dose of edoxaban on the day of the procedure.
For all patients undergoing cardioversion: Confirmation should be sought prior to cardioversion that the patient has taken edoxaban as prescribed. Decisions on initiation and duration of treatment should follow established guidelines for anticoagulant treatment in patients undergoing cardioversion.
Paediatric population
Edoxaban is not recommended for use in children and adolescents from birth to 18 years of age with confirmed VTE (PE and/or DVT) event as the efficacy has not been established.
Method of administration
For oral use.
Edoxaban can be taken with or without food.
For patients who are unable to swallow whole tablets, edoxaban tablets may be crushed and mixed with water or apple puree and immediately administered orally. Alternatively, edoxaban tablets may be crushed and suspended in a small amount of water and immediately delivered through a nasogastric tube or gastric feeding tube after which it should be flushed with water. Crushed edoxaban tablets are stable in water and apple puree for up to 4 hours.
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in the formulation.
- Clinically significant active bleeding.
- Hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
- Lesion or condition, if considered to be a significant risk for major bleeding. This may include current or recent gastrointestinal ulceration, presence of malignant neoplasms at high risk of bleeding, recent brain or spinal injury, recent brain, spinal or ophthalmic surgery, recent intracranial haemorrhage, known or suspected oesophageal varices, arteriovenous malformations, vascular aneurysms or major intraspinal or intracerebral vascular abnormalities.
- Uncontrolled severe hypertension.
- Concomitant treatment with any other anticoagulants e.g. UFH, LMWH (enoxaparin, dalteparin, etc.), heparin derivatives (fondaparinux, etc.), oral anticoagulants (warfarin, dabigatran etexilate, rivaroxaban, apixaban etc.) except under specific circumstances of switching oral anticoagulant therapy or when UFH is given at doses necessary to maintain an open central venous or arterial catheter.
- Pregnancy and breast-feeding.
4.4 Special warnings and precautions for use
Edoxaban 15 mg is not indicated as monotherapy, as it may result in decreased efficacy. It is only indicated in the process of switching from edoxaban 30 mg (patients with one or more clinical factors for increased exposure; see table 1) to VKA, together with an appropriate VKA dose.
Haemorrhagic risk
Edoxaban increases the risk of bleeding and can cause serious, potentially fatal bleeding. Edoxaban, like other anticoagulants, is recommended to be used with caution in patients with increased risk of bleeding. Edoxaban administration should be discontinued if severe haemorrhage occurs.
In the clinical studies mucosal bleedings (e.g. epistaxis, gastrointestinal, genitourinary) and anaemia were seen more frequently during long term edoxaban treatment compared with VKA treatment. Thus, in addition to adequate clinical surveillance, laboratory testing of haemoglobin/haematocrit could be of value to detect occult bleeding, as judged to be appropriate.
Several sub-groups of patients, as detailed below, are at increased risk of bleeding. These patients are to be carefully monitored for signs and symptoms of bleeding complications and anaemia after initiation of treatment. Any unexplained fall in haemoglobin or blood pressure should lead to a search for a bleeding site.
The anticoagulant effect of edoxaban cannot be reliably monitored with standard laboratory testing. A specific anticoagulant reversal agent for edoxaban is not available. Haemodialysis does not significantly contribute to edoxaban clearance.
Elderly
The co-administration of edoxaban with acetylsalicylic acid (ASA) in elderly patients should be used cautiously because of a potentially higher bleeding risk.
Renal impairment
The plasma area under the curve (AUC) for subjects with mild (CrCl > 50 - 80 mL/min), moderate (CrCl 30 - 50 mL/min) and severe (CrCl < 30 mL/min but not undergoing dialysis) renal impairment was increased by 32%, 74%, and 72%, respectively, relative to subjects with normal renal function. In patients with end stage renal disease or on dialysis, edoxaban is not recommended.
Renal function in NVAF A trend towards decreasing efficacy with increasing CrCl was observed for edoxaban compared to well-managed warfarin.
Edoxaban should be used in patients with NVAF and high CrCl only after a careful evaluation of the individual thromboembolic and bleeding risk.
Assessment of renal function: CrCl should be monitored at the beginning of the treatment in all patients and afterwards when clinically indicated.
Hepatic impairment
Edoxaban is not recommended in patients with severe hepatic impairment. Edoxaban should be used with caution in patients with mild or moderate hepatic impairment.
Patients with elevated liver enzymes (ALT/AST > 2 x ULN) or total bilirubin ≥ 1.5 x ULN were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed. Periodic hepatic monitoring is recommended for patients on edoxaban treatment beyond 1 year.
Discontinuation for surgery and other interventions
If anticoagulation must be discontinued to reduce the risk of bleeding with surgical or other procedures, edoxaban should be stopped as soon as possible and preferably at least 24 hours before the procedure.
In deciding whether a procedure should be delayed until 24 hours after the last dose of edoxaban, the increased risk of bleeding should be weighed against the urgency of the intervention. Edoxaban should be restarted after the surgical or other procedures as soon as adequate haemostasis has been established, noting that the time to onset of the edoxaban anticoagulant therapeutic effect is 1 – 2 hours. If oral medicinal products cannot be taken during or after surgical intervention, consider administering a parenteral anticoagulant and then switch to oral once daily edoxaban.
Interaction with other medicinal products affecting haemostasis
Concomitant use of medicinal products affecting haemostasis may increase the risk of bleeding. These include ASA, P2Y12 platelet inhibitors, other antithrombotic agents, fibrinolytic therapy, selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs), and chronic nonsteroidal anti-inflammatory drugs (NSAIDs).
Prosthetic heart valves and moderate to severe mitral stenosis
Edoxaban has not been studied in patients with mechanical heart valves, in patients during the first 3 months after implantation of a bioprosthetic heart valve, with or without atrial fibrillation, or in patients with moderate to severe mitral stenosis. Therefore, use of edoxaban is not recommended in these patients.
Haemodynamically unstable PE patients or patients who require thrombolysis or pulmonary embolectomy
Edoxaban is not recommended as an alternative to UFH in patients with pulmonary embolism who are haemodynamically unstable or may receive thrombolysis or pulmonary embolectomy since the safety and efficacy of edoxaban have not been established in these clinical situations.
Patients with active cancer
Efficacy and safety of edoxaban in the treatment and/or prevention of VTE in patients with active cancer have not been established.
Patients with antiphospholipid syndrome
Direct acting oral anticoagulants (DOACs) including edoxaban are not recommended for patients with a history of thrombosis who are diagnosed with antiphospholipid syndrome. In particular for patients that are triple positive (for lupus anticoagulant, anticardiolipin antibodies, and anti-beta 2-glycoprotein I antibodies), treatment with DOACs could be associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
Laboratory coagulation parameters
Although treatment with edoxaban does not require routine monitoring, the effect on anticoagulation can be estimated by a calibrated quantitative anti-Factor Xa (anti-FXa) assay which may help to inform clinical decisions in particular situations as, e.g. overdose and emergency surgery.
Edoxaban prolongs standard clotting tests such as prothrombin time (PT), INR, and activated partial thromboplastin time (aPTT) as a result of Factor Xa (FXa) inhibition. Changes observed in these clotting tests at the expected therapeutic dose are, however, small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of edoxaban.
4.5 Drug interactions
Edoxaban is predominantly absorbed in the upper gastrointestinal (GI) tract. Thus, medicinal products or disease conditions that increase gastric emptying and gut motility have the possibility of reducing edoxaban dissolution and absorption.
P-gp inhibitors
Edoxaban is a substrate for the efflux transporter P-gp. In pharmacokinetic (PK) studies, concomitant administration of edoxaban with the P-gp inhibitors ciclosporin, dronedarone, erythromycin, ketoconazole, quinidine, or verapamil resulted in increased plasma concentrations of edoxaban. Concomitant use of edoxaban with ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem requires dose reduction to 30 mg once daily. Concomitant use of edoxaban with quinidine, verapamil, or amiodarone does not require dose reduction based on clinical data.
The use of edoxaban with other P-gp inhibitors including human immunodeficiency virus (HIV) protease inhibitors has not been studied.
Edoxaban 30 mg instead of 60 mg once daily must be administered during concomitant use with the following P-gp inhibitors:
- Ciclosporin: Concurrent administration of a single dose of ciclosporin 500 mg with a single dose of edoxaban 60 mg increased edoxaban AUC and maximum serum concentration (Cmax) by 73% and 74%, respectively.
- Dronedarone: Dronedarone 400 mg twice daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 5 increased edoxaban AUC and Cmax by 85% and 46%, respectively.
- Erythromycin: Erythromycin 500 mg four times daily for 8 days with a single concomitant dose of edoxaban 60 mg on day 7 increased the edoxaban AUC and Cmax by 85% and 68%, respectively.
- Ketoconazole: Ketoconazole 400 mg once daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 4, increased edoxaban AUC and Cmax by 87% and 89%, respectively.
Edoxaban 60 mg once daily is recommended during concomitant use with the following P-gp inhibitors:
- Quinidine: Quinidine 300 mg once daily on days 1 and 4 and three times daily on days 2 and 3, with a single concomitant dose of edoxaban 60 mg on day 3, increased edoxaban AUC over 24 hours by 77% and Cmax by 85%, respectively.
- Verapamil: Verapamil 240 mg once daily for 11 days with a single concomitant dose of edoxaban 60 mg on day 10 increased the edoxaban AUC and Cmax by approximately 53%.
- Amiodarone: Co-administration of amiodarone 400 mg once daily with edoxaban 60 mg once daily increased AUC by 40% and Cmax by 66%. This was not considered clinically significant. In ENGAGE AF-TIMI 48 study in NVAF, efficacy and safety results were similar for subjects with and without concomitant amiodarone use.
P-gp inducers
Co-administration of edoxaban with the P-gp inducer rifampicin led to a decrease in mean edoxaban AUC and a shortened half-life, with possible decreases in its pharmacodynamic effects. The concomitant use of edoxaban with other P-gp inducers (e.g. phenytoin, carbamazepine, phenobarbital or St. John's Wort) may lead to reduced edoxaban plasma concentrations. Edoxaban should be used with caution when co-administered with P-gp inducers.
P-gp substrates
Digoxin
Edoxaban 60 mg once daily on days 1 to 14 with co administration of multiple daily doses of digoxin 0.25 mg twice daily (days 8 and 9) and 0.25 mg once daily (days 10 to 14) increased the Cmax of edoxaban by 17%, with no significant effect on AUC or renal clearance at steady state. When the effects of edoxaban on digoxin PK were also examined, the Cmax of digoxin increased by approximately 28% and AUC by 7%. This was not considered clinically relevant. No dose modification is necessary when edoxaban is administered with digoxin.
Anticoagulants, antiplatelet, NSAIDs and SSRIs/SNRIs
Anticoagulants
Co-administration of edoxaban with other anticoagulants is contraindicated due to increased risk of bleeding.
ASA
Co-administration of ASA (100 mg or 325 mg) and edoxaban increased bleeding time relative to either medicinal product alone. Co-administration of high dose ASA (325 mg) increased the steady state Cmax and AUC of edoxaban by 35% and 32%, respectively. The concomitant chronic use of high dose ASA (325 mg) with edoxaban is not recommended. Concomitant administration of higher doses than 100 mg ASA should only be performed under medical supervision.
In clinical studies concomitant use of ASA (low dose ≤ 100 mg/day), other antiplatelet agents, and thienopyridines was permitted and resulted in approximately a 2-fold increase in major bleeding in comparison with no concomitant use, although to a similar extent in the edoxaban and warfarin groups. Co-administration of low dose ASA (≤ 100 mg) did not affect the peak or total exposure of edoxaban either after single dose or at steady-state. Edoxaban can be co-administered with low dose ASA (≤ 100 mg/day).
Platelet inhibitors
In ENGAGE AF-TIMI 48 concomitant use of thienopyridines (e.g. clopidogrel) monotherapy was permitted and resulted in increased clinically relevant bleeding although with a lower risk of bleeding on edoxaban compared to warfarin.
There is very limited experience on the use of edoxaban with dual antiplatelet therapy or fibrinolytic agents.
NSAIDs
Co-administration of naproxen and edoxaban increased bleeding time relative to either medicinal product alone. Naproxen had no effect on the Cmax and AUC of edoxaban. In clinical studies, co-administration of NSAIDs resulted in increased clinically relevant bleeding. Chronic use of NSAIDs with edoxaban is not recommended.
SSRIs/SNRIs
As with other anticoagulants the possibility may exist that patients are at increased risk of bleeding in case of concomitant use with SSRIs or SNRIs due to their reported effect on platelets.
Effect of edoxaban on other medicinal products
Edoxaban increased the Cmax of concomitantly administered digoxin by 28%; however, the AUC was not affected. Edoxaban had no effect on the Cmax and AUC of quinidine. Edoxaban decreased the Cmax and AUC of concomitantly administered verapamil by 14% and 16%, respectively.
4.6 Use in special populations (such as pregnant woman, lactating women, paediatric patients, geriatric patients etc.)
Women of childbearing potential
Women of childbearing potential should avoid becoming pregnant during treatment with edoxaban.
Pregnancy
Safety and efficacy of edoxaban have not been established in pregnant women. Studies in animals have shown reproductive toxicity. Due to the potential reproductive toxicity, the intrinsic risk of bleeding and the evidence that edoxaban passes the placenta, Supexa OD is contraindicated during pregnancy.
Breast-feeding
Safety and efficacy of edoxaban have not been established in breast-feeding women. Data from animals indicate that edoxaban is secreted into breast milk. Therefore, Supexa OD is contraindicated during breast-feeding. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from therapy.
Fertility
No specific studies with edoxaban in human beings have been conducted to evaluate effects on fertility. In a study on male and female fertility in rats no effects were seen
4.7 Effects on ability to drive and use machines
Edoxaban has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
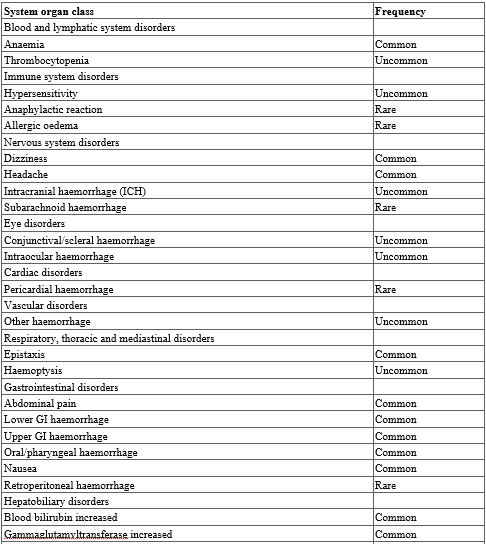
Summary of the safety profile
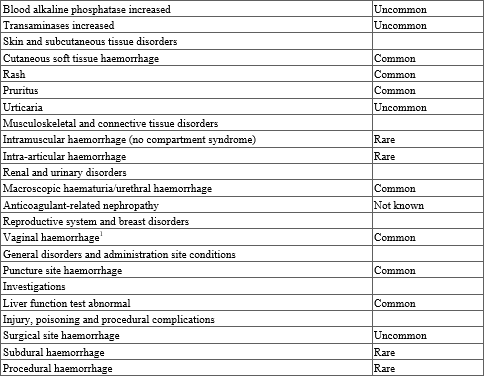
The most commonly reported adverse reactions associated with edoxaban treatment are epistaxis, haematuria and anaemia. Bleeding can occur at any site and may be severe and even fatal.
Table 3. Tabulated list of adverse reactions
Reporting of side effects
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via
email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Overdose
Overdose with Edoxaban may lead to haemorrhage. Experience with overdose cases is very limited. Studies have confirmed that Andexanet alfa, a modifed recombinant inactive form of human factor Xa that binds to and blocks the effects of factor Xa inhibitors can be used as an antidote1,2. Early administration of activated charcoal may be considered in case of Edoxaban overdose to reduce absorption. This recommendation is based on standard treatment of medicinal product overdose and data available with similar compounds, as the use of activated charcoal to reduce absorption of Edoxaban has not been specifically studied in the Edoxaban clinical programme.
Management of bleeding :
Should a bleeding complication arise in a patient receiving Edoxaban, the next Edoxaban administration should be delayed or treatment should be discontinued as appropriate. Edoxaban has a half-life of approximately 10 to 14 hours. Management should be individualized according to the severity and location of the haemorrhage. Appropriate symptomatic treatment could be used as needed, such as mechanical compression (e.g. for severe epistaxis), surgical haemostasis with bleeding control procedures, fluid replacement and haemodynamic support, blood products (packed red cells or fresh frozen plasma, depending on associated anaemia or coagulopathy) or platelets. For life-threatening bleeding that cannot be controlled with the measures such as transfusion or haemostasis, the administration of a 4-factor prothrombin complex concentrate (PCC) at 50 IU/kg has been shown to reverse the effects of Edoxaban 30 minutes after completing the infusion.
Recombinant factor VIIa (r-FVIIa) can also be considered. However, there is limited clinical experience with the use of this product in individuals receiving Edoxaban. Depending on local availability, a consultation with a coagulation expert should be considered in case of major bleedings. Protamine sulfate and vitamin K are not expected to affect the anticoagulant activity of Edoxaban. There is no experience with antifibrinolytic agents (Tranexamic acid, Aminocaproic acid) in individuals receiving Edoxaban. There is neither scientific rationale for benefit nor experience with the use of systemic haemostatics (Desmopressin, Aprotinin) in individuals receiving Edoxaban. Due to the high plasma protein binding Edoxaban is not expected to be dialyzable.
5.0 Pharmacological properties
5.1 Mechanism of action
Edoxaban is a highly selective, direct and reversible inhibitor of FXa, the serine protease located in the final common pathway of the coagulation cascade. Edoxaban inhibits free FXa, and prothrombinase activity. Inhibition of FXa in the coagulation cascade reduces thrombin generation, prolongs clotting time and reduces the risk of thrombus formation.
5.2 Pharmacodynamic properties
Edoxaban produces rapid onset of pharmacodynamic effects within 1 - 2 hours, which corresponds with peak Edoxaban exposure (Cmax). The pharmacodynamic effects measured by anti-FXa assay are predictable and correlate with the dose and the concentration of Edoxaban. As a result of FXa inhibition, Edoxaban also prolongs clotting time in tests such as PT and aPTT. Changes observed in these clotting tests are expected at the therapeutic dose, however, these changes are small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of Edoxaban.
Effects of coagulation markers when switching from Rivaroxaban, Dabigatran or Apixaban to Edoxaban : In clinical pharmacology studies, healthy subjects received Rivaroxaban 20 mg once daily, Dabigatran 150 mg twice daily or Apixaban 5 mg twice daily, followed by a single dose of Edoxaban 60 mg on day 4. The effect on PT and other coagulation biomarkers (e.g. anti-FXa, aPTT) was measured. Following the switch to Edoxaban on day 4 the PT was equivalent to day 3 of Rivaroxaban and Apixaban. For dabigatran higher aPTT activity was observed after Edoxaban administration with prior Dabigatran treatment compared to that after treatment with Edoxaban alone. This is considered to be due to the carry-over effect of dabigatran treatment, however, this did not lead to a prolongation of bleeding time.
Based on these data, when switching from these anticoagulants to Edoxaban, the first dose of Edoxaban can be initiated at the time of the next scheduled dose of the previous anticoagulant
5.3 Pharmacokinetic properties
The pharmacokinetic parameters of Edoxaban in plasma after a single dose of Supexa OD 60 were as follows.
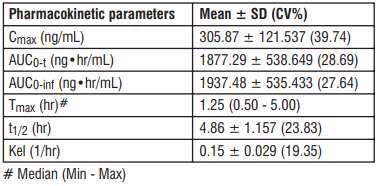
Absorption : Edoxaban is absorbed with peak plasma concentrations within 1 - 2 hours following oral administration of Edoxaban tablets. The absolute bioavailability is approximately 62%. Food increases peak exposure of Edoxaban tablets to a varying extent, but has minimal effect on total exposure.Edoxaban is poorly soluble at pH of 6.0 or higher. Co-administration of proton-pump inhibitors had no relevant impact on Edoxaban exposure.
In a study with 30 healthy subjects, both mean AUC and Cmax values for 60 mg Edoxaban administered as a crushed tablet orally mixed in apple puree or via nasogastric tube suspended in water were bioequivalent to the intact tablet. Given the predictable, dose-proportional pharmacokinetic profile of Edoxaban, the bioavailability results from this study are likely applicable to lower Edoxaban doses.
Distribution : Disposition is biphasic. The volume of distribution is 107 (19.9) L mean (SD). In vitro plasma protein binding is approximately 55%. There is no clinically relevant accumulation of Edoxaban (accumulation ratio 1.14) with once daily dosing. Steady state concentrations are achieved within 3 days.
Biotransformation : Unchanged Edoxaban is the predominant form in plasma. Edoxaban is metabolized via hydrolysis (mediated by carboxylesterase 1), conjugation or oxidation by CYP3A4/5 (< 10%). Edoxaban has three active metabolites, the predominant metabolite (M-4), formed by hydrolysis, is active and reaches less than 10% of the exposure of the parent compound in healthy subjects. Exposure to the other metabolites is less than 5%. Edoxaban is a substrate for the efflux transporter P-gp, but not a substrate for uptake transporters such as organic anion transporter polypeptide OATP1B1, organic anion transporters OAT1 or OAT3 or organic cation transporter OCT2. Its active metabolite is a substrate for OATP1B1.
Elimination : In healthy subjects, the total clearance is reported as 22 (± 3) L/hour; 50% is renally cleared (11 L/hour). Renal clearance accounts for approximately 35% of the administered dose. Metabolism and biliary / intestinal excretion account for the remaining clearance.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Acute oral toxicity study in rat and mice : A daily oral administration of Edoxaban Tosylate Monohydrate was given for 14 consecutive days to twenty-five Wistar Rats and twenty-five Swiss Albino Mice (5 per group). Each group (G1, G2, G3, G4, and G5) was administered a dose of 0, 250, 500, 1000 and 2000 mg/kg bw respectively. No mortality was observed in any dose group throughout the experiment period. All animals appeared normal throughout the study period. No external or internal gross pathological changes were observed in any of the treated animals.
Repeated dose oral toxicity study in rabbit and rat : A daily single dose of Edoxaban Tosylate Monohydrate was given for 28 days to Rabbits (3/sex/group) and Wistar Rats (6/sex/group). The selected oral doses selected were 0 (Control), 1.55, 3.1 and 6.2 mg/kg bw for Rabbit and 0 (Control), 3.1, 6.2 and 12.4 mg/kg bw. In reversal groups, animals were received control and high-dose for 28 days and observed for the next 14 days. No mortality and morbidity were observed among all the groups of animals throughout the experiment period. No clinical signs were observed in any groups of the animals throughout the experiment period. No statistically significant difference in the body weight, body weight change and feed consumption of the main study group and reversal high-dose animals compared to the control group.
Reproductive toxicology : Edoxaban showed vaginal haemorrhage at higher doses in rats and rabbits but had no effects in the reproductive performance of parent rats. In rats, no effects on male or female fertility were seen.
In animal reproduction studies, rabbits showed increased incidence of gallbladder variations at a dose of 200 mg/kg which is approximately 65 times the maximum recommended human dose (MRHD) of 60 mg/day based on total body surface area in mg/m2. Increased post-implantation pregnancy losses occurred in rats at 300 mg/kg/day (approximately 49 times the MRHD) and in rabbits at 200 mg/kg/day (approximately 65 times the MRHD) respectively. Edoxaban was excreted in the breast milk of lactating rats. Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, or phototoxicity.
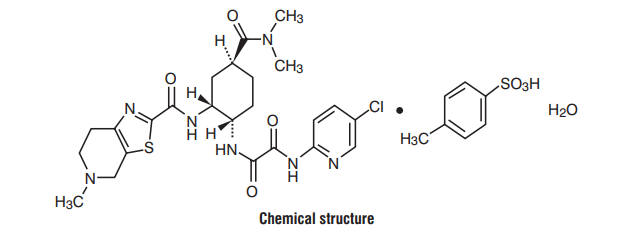
7.0 Description
Edoxaban, a factor Xa inhibitor, is supplied as Edoxaban tosylate monohydrate. The chemical name is N-(5-Chloropyridin-2-yl)- N′-[(1S,2R,4S)-4-(N, N-dimethylcarbamoyl)-2-(5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2- carboxamido)cyclohexyl] oxamide mono (4-methylbenzenesulfonate) monohydrate.
Molecular formula : C24H30ClN7O4S•C7H8O3S•H2O
Molecular weight : 738.27 g/mo
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Supexa OD 15 : Alu-Alu blister strip of 15 tablets.
Supexa OD 30 : Alu-Alu blister strip of 15 tablets.
Supexa OD 60 : Alu-Alu blister strip of 15 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- Advise patients to take Supexa OD tablet exactly as prescribed.
- Remind patients to not discontinue Supexa OD tablet without talking to the healthcare provider who prescribed it.
- Instruct patients to keep an adequate supply of tablets to ensure continuous dosing of Supexa OD tablet.
- Instruct patients who cannot swallow the tablet whole to crush Supexa OD tablet, combine with 2 to 3 ounces of water or applesauce and ingest immediately.
- Instruct patients who require a gastric tube to crush the Supexa OD tablet and mix it with 2 to 3 ounces of water before administering immediately via the gastric feeding tube.
- Inform patients that if a dose is missed, they should take Supexa OD tablet as soon as possible the same day and resume the normal dosing schedule the following day. The dose should not be doubled to make up for a missing dose.
Bleeding risk
- Advise patients that they may bleed more easily, may bleed longer or bruise more easily when treated with Supexa OD tablet.
- Instruct patients to report any unusual bleeding immediately to their healthcare provider.
- For patients that are having neuraxial anesthesia or spinal puncture, advise patients to watch for signs and symptoms of spinal or epidural hematoma, such as back pain, tingling, numbness (especially in the lower limbs), muscle weakness and stool or urine incontinence. If any of these symptoms occur, advise the patient to contact his or her physician immediately.
Invasive or surgical procedures
- Remind patients to inform their healthcare providers that they are taking Supexa OD tablet before any surgery, medical or dental procedure is scheduled.
Concomitant medication and herbals
- Remind patients to inform their healthcare providers and dentists if they plan to take or are taking any prescription medications, over-the-counter drugs or herbal products.
Pregnancy
- Remind patients to inform their healthcare provider immediately if they become pregnant or intend to become pregnant during treatment with Supexa OD tablet.
- Inform patients to not breast-feed if they are taking Supexa OD tablet.
12.0 Date of issue
06 January 2025
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Supexa OD is and what it is used for
- What you need to know before you take Supexa OD
- How to take Supexa OD
- Possible side effects
- How to store Supexa OD
- Contents of the pack and other information
1. What Supexa OD is and what it is used for
Supexa OD contains the active substance edoxaban and belongs to a group of medicines called anticoagulants. This medicine helps to prevent blood clots from forming. It works by blocking the activity of factor Xa, which is an important component of blood clotting.
Supexa OD is used in adults to:
- prevent blood clots in the brain (stroke) and other blood vessels in the body if you have a form of irregular heart rhythm called nonvalvular atrial fibrillation and at least one additional risk factor, such as heart failure, previous stroke or high blood pressure;
- treat blood clots in the veins of the legs (deep vein thrombosis) and in the blood vessels in the lungs (pulmonary embolism), and to prevent blood clots from re-occurring in the blood vessels in the legs and/or lungs.
2. What you need to know before you take Supexa OD
Do not take Supexa OD
- if you are allergic to edoxaban or any of the other ingredients of this medicine
- if you are actively bleeding
- if you have a disease or condition that increases the risk of serious bleeding (e.g. stomach ulcer, injury or bleeding in the brain, or recent surgery of the brain or eyes).
- if you are taking other medicines to prevent blood clotting (e.g. warfarin, dabigatran, rivaroxaban, apixaban or heparin), except when changing anticoagulant treatment or while getting heparin through a venous or arterial line to keep it open.
- if you have a liver disease which leads to an increased risk of bleeding.
- if you have uncontrolled high blood pressure.
- if you are pregnant or breast feeding.
Warnings and precautions
Talk to your doctor or pharmacist before taking Supexa OD,
if you have an increased risk of bleeding, as could be the case if you have any of the following conditions:
- end stage kidney disease or if you are on dialysis
- severe liver disease
- bleeding disorders
- a problem with the blood vessels in the back of your eyes (retinopathy)
- recent bleeding in your brain (intracranial or intracerebral bleeding)
- problems with the blood vessels in your brain or spinal column
If you have a mechanical heart valve.
Supexa OD 15 mg is only to be used when changing from Supexa OD 30 mg to a vitamin K antagonist (e.g. warfarin).
Take special care with Supexa OD,
- if you know that you have a disease called antiphospholipid syndrome (a disorder of the immune system that causes an increased risk for blood clots), tell your doctor who will decide if the treatment may need to be changed.
If you need to have an operation
- it is very important to take Supexa OD before and after the operation exactly at the times you have been told by your doctor. If possible, Supexa OD should be stopped at least 24 hours before an operation. Your doctor will determine when to restart Supexa OD. In emergency situations your physician will help determine the appropriate actions regarding Supexa OD.
Children and adolescents
Supexa OD is not recommended in children and adolescents under 18 years of age.
Other medicines and Supexa OD
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
If you are taking any of the following:
- some medicines for fungal infections (e.g. ketoconazole);
- medicines to treat abnormal heart beat (e.g. dronedarone, quinidine, verapamil);
- other medicines to reduce blood clotting (e.g. heparin, clopidogrel or vitamin K antagonists such as warfarin, acenocoumarol, phenprocoumon or dabigatran, rivaroxaban, apixaban);
- antibiotic medicines (e.g. erythromycin, clarithromycin);
- medicines to prevent organ rejection after transplantation (e.g. ciclosporin);
- anti-inflammatory and pain-relieving medicines (e.g. naproxen or acetylsalicylic acid);
- antidepressant medicines called selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors;
If any of the above apply to you, tell your doctor before taking Supexa OD, because these medicines may increase the effects of Supexa OD and the chance of unwanted bleeding. Your doctor will decide, if you should be treated with Supexa OD and if you should be kept under observation.
If you are taking any of the following:
- some medicines for treatment of epilepsy (e.g. phenytoin, carbamazepine, phenobarbital).
- St John’s Wort, a herbal product used for anxiety and mild depression.
- rifampicin, an antibiotic medicine.
If any of the above apply to you, tell your doctor before taking Supexa OD, because the effect of Supexa OD may be reduced. Your doctor will decide if you should be treated with Supexa OD and if you should be kept under observation.
Pregnancy and breast-feeding
Do not take Supexa OD if you are pregnant or breast-feeding. If there is a chance that you could become pregnant, use a reliable contraceptive while you are taking Supexa OD. If you become pregnant while you are taking Supexa OD, immediately tell your doctor, who will decide how you should be treated.
Driving and using machines
Supexa OD has no or negligible effects on your ability to drive or use machines.
3. How to take Supexa OD
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
How much to take?
The recommended dose is one 60 mg tablet once daily.
If you have impaired kidney function, the dose may be reduced to one 30 mg tablet once daily by your doctor.
if your body weight is 60 kg or lower, the recommended dose is one 30 mg tablet once daily;
if your doctor has prescribed medicines known as P-gp inhibitors: ciclosporin, dronedarone, erythromycin, or ketoconazole, the recommended dose is one 30 mg tablet once daily.
How to take the tablet?
Swallow the tablet, preferably with water.
Supexa OD can be taken with or without food.
If you have difficulty swallowing the tablet whole, talk to your doctor about other ways to take Supexa OD. The tablet may be crushed and mixed with water or apple puree immediately before you take it. If necessary, your doctor may also give you the crushed Supexa OD tablet through a tube via the nose (nasogastric tube) or a tube in the stomach (gastric feeding tube).
Your doctor may change your anticoagulant treatment as follows:
Changing from vitamin K antagonists (e.g. warfarin) to Supexa OD
Stop taking the vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to start taking Supexa OD.
Changing from non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban) to Supexa OD
Stop taking the previous medicines (e.g. dabigatran, rivaroxaban, or apixaban) and start Supexa OD at the time of the next scheduled dose.
Changing from parenteral anticoagulants (e.g. heparin) to Supexa OD
Stop taking the anticoagulant (e.g. heparin) and start Supexa OD at the time of the next scheduled anticoagulant dose.
Changing from Supexa OD to vitamin K antagonists (e.g. warfarin)
If you currently take 60 mg Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 30 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
If you currently take 30 mg (dose reduced) Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 15 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
Changing from Supexa OD to non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban)
Stop taking Supexa OD and start the non-VKA anticoagulant (e.g. dabigatran, rivaroxaban, or apixaban) at the time of the next scheduled dose of Supexa OD.
Changing from Supexa OD to parenteral anticoagulants (e.g. heparin)
Stop taking Supexa OD and start the parenteral anticoagulant (e.g. heparin) at the time of the next scheduled dose of Supexa OD.
Patients undergoing cardioversion:
If your abnormal heartbeat needs to be restored to normal by a procedure called cardioversion, take Supexa OD at the times your doctor tells you to prevent blood clots in the brain and other blood vessels in your body.
If you take more Supexa OD than you should
Tell your doctor immediately if you have taken too many Supexa OD tablets.
If you take more Supexa OD than recommended, you may have an increased risk of bleeding.
If you forget to take Supexa OD
You should take the tablet immediately and then continue the following day with the once daily tablet as usual. Do not take a double dose on the same day to make up for a forgotten dose.
If you stop taking Supexa OD
Do not stop taking Supexa OD without talking to your doctor first, because Supexa OD treats and prevents serious conditions.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them. Like other similar medicines (medicines to reduce blood clotting), Supexa OD may cause bleeding which may potentially be life-threatening. In some cases, the bleeding may not be obvious.
If you experience any bleeding event that does not stop by itself or if you experience signs of excessive bleeding (exceptional weakness, tiredness, paleness, dizziness, headache or unexplained swelling) consult your doctor immediately.
Your doctor may decide to keep you under closer observation or change your medicine.
Overall list of possible side effects:
Common (may affect up to 1 in 10 people)
- stomach ache
- abnormal liver blood tests
- bleeding from the skin or under the skin
- anaemia (low levels of red blood cells)
- bleeding from the nose
- bleeding from the vagina
- rash
- bleeding in the bowel
- bleeding from the mouth and/or throat
- blood found in your urine
- bleeding following an injury (puncture)
- bleeding in the stomach
- dizziness
- feeling sick
- headache
- itching
Uncommon (may affect up to 1 in 100 people)
- bleeding in the eyes
- bleeding from a surgical wound following an operation
- blood in the spit when coughing
- bleeding in the brain
- other types of bleeding
- reduced number of platelets in your blood (which can affect clotting)
- allergic reaction
- hives
Rare (may affect up to 1 in 1,000 people)
- bleeding in the muscles
- bleeding in joints
- bleeding in the abdomen
- bleeding in the heart
- bleeding inside the skull
- bleeding following a surgical procedure
- allergic shock
- swelling of any part of the body due to allergic reaction
Not known (frequency cannot be estimated from the available data)
- bleeding in the kidney sometimes with presence of blood in urine leading to inability of the kidneys to work properly (anticoagulant-related nephropathy).
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Supexa OD
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the carton and on each blister or bottle after EXP. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Supexa OD contains
- The active substance is edoxaban (as tosylate monohydrate).
Supexa OD 15 mg film-coated tablets
Each tablet contains 15 mg edoxaban (as tosylate monohydrate).
Supexa OD 30 mg film-coated tablets
Each tablet contains 30 mg edoxaban (as tosylate monohydrate).
Supexa OD 60 mg film-coated tablets
Each tablet contains 60 mg edoxaban (as tosylate monohydrate).
For More Information About This Product
Supexa- OD 30 Tablets
1.0 Generic name
Edoxaban Tablets
2.0 Qualitative and quantitative composition
SUPEXA OD 15
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….15 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 30
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….30 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 60
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….60 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
3.0 Dosage form and strength
Film coated tablet, 15 mg / 30 mg / 60 mg
4.0 Clinical particulars
4.1Therapeutic indications
- For prevention of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation (NVAF) with one or more risk factors, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischaemic attack (TIA).
- For the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the prevention of recurrent DVT and PE in adults.
4.2 Posology and method of administration
Posology
Prevention of stroke and systemic embolism
- The recommended dose is 60 mg edoxaban once daily. Therapy with edoxaban in NVAF patients should be continued long term.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate or severe renal impairment (creatinine clearance (CrCl) 15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of the following P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- For elderly patients aged 80 years and above: dose reduced to 15 mg once daily.
Treatment of DVT, PE and prevention of recurrent DVT and PE (venous thromboembolism (VTE))
- The recommended dose is 60 mg edoxaban once daily following initial use of parenteral anticoagulant for at least 5 days. Edoxaban and initial parenteral anticoagulant should not be administered simultaneously. The duration of therapy for treatment of DVT and PE (VTE), and prevention of recurrent VTE should be individualized after careful assessment of the treatment benefit against the risk for bleeding. Short duration of therapy (at least 3 months) should be based on transient risk factors (e.g. recent surgery, trauma, immobilization) and longer durations should be based on permanent risk factors or idiopathic DVT or PE.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate to severe renal impairment (CrCl -15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Prevention of venous thromboembolism (VTE) in patients undergoing orthopedic surgery for the lower limbs - Total knee or hip replacement, hip fracture surgery:
- The usual adult dosage is 30 mg of edoxaban administered orally once daily.
- Consider reducing the dose to 15 mg once daily if
- Renal impairment (CrCl 30 mL/min and ≤ 50 mL/min).
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Edoxaban should be administered 12 hours after surgery and after confirming that there is no bleeding from the surgical wound, etc.
- Edoxaban should be at least 2 hours after the epidural catheter is removed or lumbar puncture is performed.
Table 1: Summary of Edoxaban Posology in NVAF and VTE (DVT and PE)
Missed dose
If a dose of edoxaban is missed, the dose should be taken immediately and then be continued the following day with the once-daily intake as recommended. The patient should not take double the prescribed dose on the same day to make up for a missed dose. Switching to and from edoxaban Continued anticoagulant therapy is important in patients with NVAF and VTE. There may be situations that warrant a change in anticoagulation therapy (Table 2).
Table 2: Switching of anticoagulant treatment in NVAF and VTE (DVT and PE)
Special populations
Elderly population
No dose reduction is required.
Renal impairment
Renal function should be assessed in all patients by calculating the CrCl prior to initiation of treatment with edoxaban to exclude patients with end stage renal disease (i.e. CrCl < 15 mL/min), to use the correct edoxaban dose in patients with CrCl 15 – 50 mL/min (30 mg once daily), in patients with CrCl > 50 mL/min (60 mg once daily) and when deciding on the use of edoxaban in patients with increased CrCl.
Renal function should also be assessed when a change in renal function is suspected during treatment (e.g. hypovolaemia, dehydration, and in case of concomitant use of certain medicinal products).
The method used to estimate renal function (CrCl in mL/min) during the clinical development of edoxaban was the Cockcroft-Gault method. The formula is as follows:
For creatinine in µ mol/L:
1.23× (140-age [years])× weight [kg] (× 0.85 if Female)
Serum creatinine [µmol/L]
For creatinine in mg/dL:
(140-age [years]) × weight [kg] (× 0.85 if Female)
72×Serum creatinine [mg/dL]
This method is recommended when assessing patients' CrCl prior to and during edoxaban treatment.
In patients with mild renal impairment (CrCl > 50 – 80 mL/min), the recommended dose is 60 mg edoxaban once daily.
In patients with moderate or severe renal impairment (CrCl 15 – 50 mL/min), the recommended dose is 30 mg edoxaban once daily.
In patients with end stage renal disease (ESRD) (CrCl < 15 mL/min) or on dialysis, the use of edoxaban is not recommended.
Hepatic impairment
Edoxaban is contraindicated in patients with hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
In patients with severe hepatic impairment edoxaban is not recommended.
In patients with mild to moderate hepatic impairment the recommended dose is 60 mg edoxaban once daily. Edoxaban should be used with caution in patients with mild to moderate hepatic impairment.
Patients with elevated liver enzymes (alanine aminotransferase (ALT) or aspartate transaminase (AST) > 2 x upper limit of normal (ULN)) or total bilirubin ≥ 1.5 x ULN, were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed.
Body weight
For patients with body weight ≤ 60 kg, the recommended dose is 30 mg edoxaban once daily).
Gender
No dose reduction is required.
Concomitant use of edoxaban with P-glycoprotein (P-gp) inhibitors
In patients concomitantly taking edoxaban and the following P-gp inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem, the recommended dose is 30 mg edoxaban once daily.
No dose reduction is required for concomitant use of amiodarone, quinidine or verapamil. The use of edoxaban with other P-gp inhibitors including HIV protease inhibitors has not been studied.
Patients undergoing cardioversion
Edoxaban can be initiated or continued in patients who may require cardioversion. For transoesophageal echocardiogram (TEE) guided cardioversion in patients not previously treated with anticoagulants, edoxaban treatment should be started at least 2 hours before cardioversion to ensure adequate anticoagulation. Cardioversion should be performed no later than 12 hours after the dose of edoxaban on the day of the procedure.
For all patients undergoing cardioversion: Confirmation should be sought prior to cardioversion that the patient has taken edoxaban as prescribed. Decisions on initiation and duration of treatment should follow established guidelines for anticoagulant treatment in patients undergoing cardioversion.
Paediatric population
Edoxaban is not recommended for use in children and adolescents from birth to 18 years of age with confirmed VTE (PE and/or DVT) event as the efficacy has not been established.
Method of administration
For oral use.
Edoxaban can be taken with or without food.
For patients who are unable to swallow whole tablets, edoxaban tablets may be crushed and mixed with water or apple puree and immediately administered orally. Alternatively, edoxaban tablets may be crushed and suspended in a small amount of water and immediately delivered through a nasogastric tube or gastric feeding tube after which it should be flushed with water. Crushed edoxaban tablets are stable in water and apple puree for up to 4 hours.
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in the formulation.
- Clinically significant active bleeding.
- Hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
- Lesion or condition, if considered to be a significant risk for major bleeding. This may include current or recent gastrointestinal ulceration, presence of malignant neoplasms at high risk of bleeding, recent brain or spinal injury, recent brain, spinal or ophthalmic surgery, recent intracranial haemorrhage, known or suspected oesophageal varices, arteriovenous malformations, vascular aneurysms or major intraspinal or intracerebral vascular abnormalities.
- Uncontrolled severe hypertension.
- Concomitant treatment with any other anticoagulants e.g. UFH, LMWH (enoxaparin, dalteparin, etc.), heparin derivatives (fondaparinux, etc.), oral anticoagulants (warfarin, dabigatran etexilate, rivaroxaban, apixaban etc.) except under specific circumstances of switching oral anticoagulant therapy or when UFH is given at doses necessary to maintain an open central venous or arterial catheter.
- Pregnancy and breast-feeding.
4.4 Special warnings and precautions for use
Edoxaban 15 mg is not indicated as monotherapy, as it may result in decreased efficacy. It is only indicated in the process of switching from edoxaban 30 mg (patients with one or more clinical factors for increased exposure; see table 1) to VKA, together with an appropriate VKA dose.
Haemorrhagic risk
Edoxaban increases the risk of bleeding and can cause serious, potentially fatal bleeding. Edoxaban, like other anticoagulants, is recommended to be used with caution in patients with increased risk of bleeding. Edoxaban administration should be discontinued if severe haemorrhage occurs.
In the clinical studies mucosal bleedings (e.g. epistaxis, gastrointestinal, genitourinary) and anaemia were seen more frequently during long term edoxaban treatment compared with VKA treatment. Thus, in addition to adequate clinical surveillance, laboratory testing of haemoglobin/haematocrit could be of value to detect occult bleeding, as judged to be appropriate.
Several sub-groups of patients, as detailed below, are at increased risk of bleeding. These patients are to be carefully monitored for signs and symptoms of bleeding complications and anaemia after initiation of treatment. Any unexplained fall in haemoglobin or blood pressure should lead to a search for a bleeding site.
The anticoagulant effect of edoxaban cannot be reliably monitored with standard laboratory testing. A specific anticoagulant reversal agent for edoxaban is not available. Haemodialysis does not significantly contribute to edoxaban clearance.
Elderly
The co-administration of edoxaban with acetylsalicylic acid (ASA) in elderly patients should be used cautiously because of a potentially higher bleeding risk.
Renal impairment
The plasma area under the curve (AUC) for subjects with mild (CrCl > 50 - 80 mL/min), moderate (CrCl 30 - 50 mL/min) and severe (CrCl < 30 mL/min but not undergoing dialysis) renal impairment was increased by 32%, 74%, and 72%, respectively, relative to subjects with normal renal function. In patients with end stage renal disease or on dialysis, edoxaban is not recommended.
Renal function in NVAF A trend towards decreasing efficacy with increasing CrCl was observed for edoxaban compared to well-managed warfarin.
Edoxaban should be used in patients with NVAF and high CrCl only after a careful evaluation of the individual thromboembolic and bleeding risk.
Assessment of renal function: CrCl should be monitored at the beginning of the treatment in all patients and afterwards when clinically indicated.
Hepatic impairment
Edoxaban is not recommended in patients with severe hepatic impairment. Edoxaban should be used with caution in patients with mild or moderate hepatic impairment.
Patients with elevated liver enzymes (ALT/AST > 2 x ULN) or total bilirubin ≥ 1.5 x ULN were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed. Periodic hepatic monitoring is recommended for patients on edoxaban treatment beyond 1 year.
Discontinuation for surgery and other interventions
If anticoagulation must be discontinued to reduce the risk of bleeding with surgical or other procedures, edoxaban should be stopped as soon as possible and preferably at least 24 hours before the procedure.
In deciding whether a procedure should be delayed until 24 hours after the last dose of edoxaban, the increased risk of bleeding should be weighed against the urgency of the intervention. Edoxaban should be restarted after the surgical or other procedures as soon as adequate haemostasis has been established, noting that the time to onset of the edoxaban anticoagulant therapeutic effect is 1 – 2 hours. If oral medicinal products cannot be taken during or after surgical intervention, consider administering a parenteral anticoagulant and then switch to oral once daily edoxaban.
Interaction with other medicinal products affecting haemostasis
Concomitant use of medicinal products affecting haemostasis may increase the risk of bleeding. These include ASA, P2Y12 platelet inhibitors, other antithrombotic agents, fibrinolytic therapy, selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs), and chronic nonsteroidal anti-inflammatory drugs (NSAIDs).
Prosthetic heart valves and moderate to severe mitral stenosis
Edoxaban has not been studied in patients with mechanical heart valves, in patients during the first 3 months after implantation of a bioprosthetic heart valve, with or without atrial fibrillation, or in patients with moderate to severe mitral stenosis. Therefore, use of edoxaban is not recommended in these patients.
Haemodynamically unstable PE patients or patients who require thrombolysis or pulmonary embolectomy
Edoxaban is not recommended as an alternative to UFH in patients with pulmonary embolism who are haemodynamically unstable or may receive thrombolysis or pulmonary embolectomy since the safety and efficacy of edoxaban have not been established in these clinical situations.
Patients with active cancer
Efficacy and safety of edoxaban in the treatment and/or prevention of VTE in patients with active cancer have not been established.
Patients with antiphospholipid syndrome
Direct acting oral anticoagulants (DOACs) including edoxaban are not recommended for patients with a history of thrombosis who are diagnosed with antiphospholipid syndrome. In particular for patients that are triple positive (for lupus anticoagulant, anticardiolipin antibodies, and anti-beta 2-glycoprotein I antibodies), treatment with DOACs could be associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
Laboratory coagulation parameters
Although treatment with edoxaban does not require routine monitoring, the effect on anticoagulation can be estimated by a calibrated quantitative anti-Factor Xa (anti-FXa) assay which may help to inform clinical decisions in particular situations as, e.g. overdose and emergency surgery.
Edoxaban prolongs standard clotting tests such as prothrombin time (PT), INR, and activated partial thromboplastin time (aPTT) as a result of Factor Xa (FXa) inhibition. Changes observed in these clotting tests at the expected therapeutic dose are, however, small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of edoxaban.
4.5 Drug interactions
Edoxaban is predominantly absorbed in the upper gastrointestinal (GI) tract. Thus, medicinal products or disease conditions that increase gastric emptying and gut motility have the possibility of reducing edoxaban dissolution and absorption.
P-gp inhibitors
Edoxaban is a substrate for the efflux transporter P-gp. In pharmacokinetic (PK) studies, concomitant administration of edoxaban with the P-gp inhibitors ciclosporin, dronedarone, erythromycin, ketoconazole, quinidine, or verapamil resulted in increased plasma concentrations of edoxaban. Concomitant use of edoxaban with ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem requires dose reduction to 30 mg once daily. Concomitant use of edoxaban with quinidine, verapamil, or amiodarone does not require dose reduction based on clinical data.
The use of edoxaban with other P-gp inhibitors including human immunodeficiency virus (HIV) protease inhibitors has not been studied.
Edoxaban 30 mg instead of 60 mg once daily must be administered during concomitant use with the following P-gp inhibitors:
- Ciclosporin: Concurrent administration of a single dose of ciclosporin 500 mg with a single dose of edoxaban 60 mg increased edoxaban AUC and maximum serum concentration (Cmax) by 73% and 74%, respectively.
- Dronedarone: Dronedarone 400 mg twice daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 5 increased edoxaban AUC and Cmax by 85% and 46%, respectively.
- Erythromycin: Erythromycin 500 mg four times daily for 8 days with a single concomitant dose of edoxaban 60 mg on day 7 increased the edoxaban AUC and Cmax by 85% and 68%, respectively.
- Ketoconazole: Ketoconazole 400 mg once daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 4, increased edoxaban AUC and Cmax by 87% and 89%, respectively.
Edoxaban 60 mg once daily is recommended during concomitant use with the following P-gp inhibitors:
- Quinidine: Quinidine 300 mg once daily on days 1 and 4 and three times daily on days 2 and 3, with a single concomitant dose of edoxaban 60 mg on day 3, increased edoxaban AUC over 24 hours by 77% and Cmax by 85%, respectively.
- Verapamil: Verapamil 240 mg once daily for 11 days with a single concomitant dose of edoxaban 60 mg on day 10 increased the edoxaban AUC and Cmax by approximately 53%.
- Amiodarone: Co-administration of amiodarone 400 mg once daily with edoxaban 60 mg once daily increased AUC by 40% and Cmax by 66%. This was not considered clinically significant. In ENGAGE AF-TIMI 48 study in NVAF, efficacy and safety results were similar for subjects with and without concomitant amiodarone use.
P-gp inducers
Co-administration of edoxaban with the P-gp inducer rifampicin led to a decrease in mean edoxaban AUC and a shortened half-life, with possible decreases in its pharmacodynamic effects. The concomitant use of edoxaban with other P-gp inducers (e.g. phenytoin, carbamazepine, phenobarbital or St. John's Wort) may lead to reduced edoxaban plasma concentrations. Edoxaban should be used with caution when co-administered with P-gp inducers.
P-gp substrates
Digoxin
Edoxaban 60 mg once daily on days 1 to 14 with co administration of multiple daily doses of digoxin 0.25 mg twice daily (days 8 and 9) and 0.25 mg once daily (days 10 to 14) increased the Cmax of edoxaban by 17%, with no significant effect on AUC or renal clearance at steady state. When the effects of edoxaban on digoxin PK were also examined, the Cmax of digoxin increased by approximately 28% and AUC by 7%. This was not considered clinically relevant. No dose modification is necessary when edoxaban is administered with digoxin.
Anticoagulants, antiplatelet, NSAIDs and SSRIs/SNRIs
Anticoagulants
Co-administration of edoxaban with other anticoagulants is contraindicated due to increased risk of bleeding.
ASA
Co-administration of ASA (100 mg or 325 mg) and edoxaban increased bleeding time relative to either medicinal product alone. Co-administration of high dose ASA (325 mg) increased the steady state Cmax and AUC of edoxaban by 35% and 32%, respectively. The concomitant chronic use of high dose ASA (325 mg) with edoxaban is not recommended. Concomitant administration of higher doses than 100 mg ASA should only be performed under medical supervision.
In clinical studies concomitant use of ASA (low dose ≤ 100 mg/day), other antiplatelet agents, and thienopyridines was permitted and resulted in approximately a 2-fold increase in major bleeding in comparison with no concomitant use, although to a similar extent in the edoxaban and warfarin groups. Co-administration of low dose ASA (≤ 100 mg) did not affect the peak or total exposure of edoxaban either after single dose or at steady-state. Edoxaban can be co-administered with low dose ASA (≤ 100 mg/day).
Platelet inhibitors
In ENGAGE AF-TIMI 48 concomitant use of thienopyridines (e.g. clopidogrel) monotherapy was permitted and resulted in increased clinically relevant bleeding although with a lower risk of bleeding on edoxaban compared to warfarin.
There is very limited experience on the use of edoxaban with dual antiplatelet therapy or fibrinolytic agents.
NSAIDs
Co-administration of naproxen and edoxaban increased bleeding time relative to either medicinal product alone. Naproxen had no effect on the Cmax and AUC of edoxaban. In clinical studies, co-administration of NSAIDs resulted in increased clinically relevant bleeding. Chronic use of NSAIDs with edoxaban is not recommended.
SSRIs/SNRIs
As with other anticoagulants the possibility may exist that patients are at increased risk of bleeding in case of concomitant use with SSRIs or SNRIs due to their reported effect on platelets.
Effect of edoxaban on other medicinal products
Edoxaban increased the Cmax of concomitantly administered digoxin by 28%; however, the AUC was not affected. Edoxaban had no effect on the Cmax and AUC of quinidine. Edoxaban decreased the Cmax and AUC of concomitantly administered verapamil by 14% and 16%, respectively.
4.6 Use in special populations (such as pregnant woman, lactating women, paediatric patients, geriatric patients etc.)
Women of childbearing potential
Women of childbearing potential should avoid becoming pregnant during treatment with edoxaban.
Pregnancy
Safety and efficacy of edoxaban have not been established in pregnant women. Studies in animals have shown reproductive toxicity. Due to the potential reproductive toxicity, the intrinsic risk of bleeding and the evidence that edoxaban passes the placenta, Supexa OD is contraindicated during pregnancy.
Breast-feeding
Safety and efficacy of edoxaban have not been established in breast-feeding women. Data from animals indicate that edoxaban is secreted into breast milk. Therefore, Supexa OD is contraindicated during breast-feeding. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from therapy.
Fertility
No specific studies with edoxaban in human beings have been conducted to evaluate effects on fertility. In a study on male and female fertility in rats no effects were seen
4.7 Effects on ability to drive and use machines
Edoxaban has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The most commonly reported adverse reactions associated with edoxaban treatment are epistaxis, haematuria and anaemia. Bleeding can occur at any site and may be severe and even fatal.
Table 3. Tabulated list of adverse reactions
Reporting of side effects
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via
email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Overdose
Overdose with Edoxaban may lead to haemorrhage. Experience with overdose cases is very limited. Studies have confirmed that Andexanet alfa, a modifed recombinant inactive form of human factor Xa that binds to and blocks the effects of factor Xa inhibitors can be used as an antidote1,2. Early administration of activated charcoal may be considered in case of Edoxaban overdose to reduce absorption. This recommendation is based on standard treatment of medicinal product overdose and data available with similar compounds, as the use of activated charcoal to reduce absorption of Edoxaban has not been specifically studied in the Edoxaban clinical programme.
Management of bleeding :
Should a bleeding complication arise in a patient receiving Edoxaban, the next Edoxaban administration should be delayed or treatment should be discontinued as appropriate. Edoxaban has a half-life of approximately 10 to 14 hours. Management should be individualized according to the severity and location of the haemorrhage. Appropriate symptomatic treatment could be used as needed, such as mechanical compression (e.g. for severe epistaxis), surgical haemostasis with bleeding control procedures, fluid replacement and haemodynamic support, blood products (packed red cells or fresh frozen plasma, depending on associated anaemia or coagulopathy) or platelets. For life-threatening bleeding that cannot be controlled with the measures such as transfusion or haemostasis, the administration of a 4-factor prothrombin complex concentrate (PCC) at 50 IU/kg has been shown to reverse the effects of Edoxaban 30 minutes after completing the infusion.
Recombinant factor VIIa (r-FVIIa) can also be considered. However, there is limited clinical experience with the use of this product in individuals receiving Edoxaban. Depending on local availability, a consultation with a coagulation expert should be considered in case of major bleedings. Protamine sulfate and vitamin K are not expected to affect the anticoagulant activity of Edoxaban. There is no experience with antifibrinolytic agents (Tranexamic acid, Aminocaproic acid) in individuals receiving Edoxaban. There is neither scientific rationale for benefit nor experience with the use of systemic haemostatics (Desmopressin, Aprotinin) in individuals receiving Edoxaban. Due to the high plasma protein binding Edoxaban is not expected to be dialyzable.
5.0 Pharmacological properties
5.1 Mechanism of action
Edoxaban is a highly selective, direct and reversible inhibitor of FXa, the serine protease located in the final common pathway of the coagulation cascade. Edoxaban inhibits free FXa, and prothrombinase activity. Inhibition of FXa in the coagulation cascade reduces thrombin generation, prolongs clotting time and reduces the risk of thrombus formation.
5.2 Pharmacodynamic properties
Edoxaban produces rapid onset of pharmacodynamic effects within 1 - 2 hours, which corresponds with peak Edoxaban exposure (Cmax). The pharmacodynamic effects measured by anti-FXa assay are predictable and correlate with the dose and the concentration of Edoxaban. As a result of FXa inhibition, Edoxaban also prolongs clotting time in tests such as PT and aPTT. Changes observed in these clotting tests are expected at the therapeutic dose, however, these changes are small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of Edoxaban.
Effects of coagulation markers when switching from Rivaroxaban, Dabigatran or Apixaban to Edoxaban : In clinical pharmacology studies, healthy subjects received Rivaroxaban 20 mg once daily, Dabigatran 150 mg twice daily or Apixaban 5 mg twice daily, followed by a single dose of Edoxaban 60 mg on day 4. The effect on PT and other coagulation biomarkers (e.g. anti-FXa, aPTT) was measured. Following the switch to Edoxaban on day 4 the PT was equivalent to day 3 of Rivaroxaban and Apixaban. For dabigatran higher aPTT activity was observed after Edoxaban administration with prior Dabigatran treatment compared to that after treatment with Edoxaban alone. This is considered to be due to the carry-over effect of dabigatran treatment, however, this did not lead to a prolongation of bleeding time.
Based on these data, when switching from these anticoagulants to Edoxaban, the first dose of Edoxaban can be initiated at the time of the next scheduled dose of the previous anticoagulant
5.3 Pharmacokinetic properties
The pharmacokinetic parameters of Edoxaban in plasma after a single dose of Supexa OD 60 were as follows.
Absorption : Edoxaban is absorbed with peak plasma concentrations within 1 - 2 hours following oral administration of Edoxaban tablets. The absolute bioavailability is approximately 62%. Food increases peak exposure of Edoxaban tablets to a varying extent, but has minimal effect on total exposure.Edoxaban is poorly soluble at pH of 6.0 or higher. Co-administration of proton-pump inhibitors had no relevant impact on Edoxaban exposure.
In a study with 30 healthy subjects, both mean AUC and Cmax values for 60 mg Edoxaban administered as a crushed tablet orally mixed in apple puree or via nasogastric tube suspended in water were bioequivalent to the intact tablet. Given the predictable, dose-proportional pharmacokinetic profile of Edoxaban, the bioavailability results from this study are likely applicable to lower Edoxaban doses.
Distribution : Disposition is biphasic. The volume of distribution is 107 (19.9) L mean (SD). In vitro plasma protein binding is approximately 55%. There is no clinically relevant accumulation of Edoxaban (accumulation ratio 1.14) with once daily dosing. Steady state concentrations are achieved within 3 days.
Biotransformation : Unchanged Edoxaban is the predominant form in plasma. Edoxaban is metabolized via hydrolysis (mediated by carboxylesterase 1), conjugation or oxidation by CYP3A4/5 (< 10%). Edoxaban has three active metabolites, the predominant metabolite (M-4), formed by hydrolysis, is active and reaches less than 10% of the exposure of the parent compound in healthy subjects. Exposure to the other metabolites is less than 5%. Edoxaban is a substrate for the efflux transporter P-gp, but not a substrate for uptake transporters such as organic anion transporter polypeptide OATP1B1, organic anion transporters OAT1 or OAT3 or organic cation transporter OCT2. Its active metabolite is a substrate for OATP1B1.
Elimination : In healthy subjects, the total clearance is reported as 22 (± 3) L/hour; 50% is renally cleared (11 L/hour). Renal clearance accounts for approximately 35% of the administered dose. Metabolism and biliary / intestinal excretion account for the remaining clearance.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Acute oral toxicity study in rat and mice : A daily oral administration of Edoxaban Tosylate Monohydrate was given for 14 consecutive days to twenty-five Wistar Rats and twenty-five Swiss Albino Mice (5 per group). Each group (G1, G2, G3, G4, and G5) was administered a dose of 0, 250, 500, 1000 and 2000 mg/kg bw respectively. No mortality was observed in any dose group throughout the experiment period. All animals appeared normal throughout the study period. No external or internal gross pathological changes were observed in any of the treated animals.
Repeated dose oral toxicity study in rabbit and rat : A daily single dose of Edoxaban Tosylate Monohydrate was given for 28 days to Rabbits (3/sex/group) and Wistar Rats (6/sex/group). The selected oral doses selected were 0 (Control), 1.55, 3.1 and 6.2 mg/kg bw for Rabbit and 0 (Control), 3.1, 6.2 and 12.4 mg/kg bw. In reversal groups, animals were received control and high-dose for 28 days and observed for the next 14 days. No mortality and morbidity were observed among all the groups of animals throughout the experiment period. No clinical signs were observed in any groups of the animals throughout the experiment period. No statistically significant difference in the body weight, body weight change and feed consumption of the main study group and reversal high-dose animals compared to the control group.
Reproductive toxicology : Edoxaban showed vaginal haemorrhage at higher doses in rats and rabbits but had no effects in the reproductive performance of parent rats. In rats, no effects on male or female fertility were seen.
In animal reproduction studies, rabbits showed increased incidence of gallbladder variations at a dose of 200 mg/kg which is approximately 65 times the maximum recommended human dose (MRHD) of 60 mg/day based on total body surface area in mg/m2. Increased post-implantation pregnancy losses occurred in rats at 300 mg/kg/day (approximately 49 times the MRHD) and in rabbits at 200 mg/kg/day (approximately 65 times the MRHD) respectively. Edoxaban was excreted in the breast milk of lactating rats. Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, or phototoxicity.
7.0 Description
Edoxaban, a factor Xa inhibitor, is supplied as Edoxaban tosylate monohydrate. The chemical name is N-(5-Chloropyridin-2-yl)- N′-[(1S,2R,4S)-4-(N, N-dimethylcarbamoyl)-2-(5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2- carboxamido)cyclohexyl] oxamide mono (4-methylbenzenesulfonate) monohydrate.
Molecular formula : C24H30ClN7O4S•C7H8O3S•H2O
Molecular weight : 738.27 g/mo
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Supexa OD 15 : Alu-Alu blister strip of 15 tablets.
Supexa OD 30 : Alu-Alu blister strip of 15 tablets.
Supexa OD 60 : Alu-Alu blister strip of 15 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- Advise patients to take Supexa OD tablet exactly as prescribed.
- Remind patients to not discontinue Supexa OD tablet without talking to the healthcare provider who prescribed it.
- Instruct patients to keep an adequate supply of tablets to ensure continuous dosing of Supexa OD tablet.
- Instruct patients who cannot swallow the tablet whole to crush Supexa OD tablet, combine with 2 to 3 ounces of water or applesauce and ingest immediately.
- Instruct patients who require a gastric tube to crush the Supexa OD tablet and mix it with 2 to 3 ounces of water before administering immediately via the gastric feeding tube.
- Inform patients that if a dose is missed, they should take Supexa OD tablet as soon as possible the same day and resume the normal dosing schedule the following day. The dose should not be doubled to make up for a missing dose.
Bleeding risk
- Advise patients that they may bleed more easily, may bleed longer or bruise more easily when treated with Supexa OD tablet.
- Instruct patients to report any unusual bleeding immediately to their healthcare provider.
- For patients that are having neuraxial anesthesia or spinal puncture, advise patients to watch for signs and symptoms of spinal or epidural hematoma, such as back pain, tingling, numbness (especially in the lower limbs), muscle weakness and stool or urine incontinence. If any of these symptoms occur, advise the patient to contact his or her physician immediately.
Invasive or surgical procedures
- Remind patients to inform their healthcare providers that they are taking Supexa OD tablet before any surgery, medical or dental procedure is scheduled.
Concomitant medication and herbals
- Remind patients to inform their healthcare providers and dentists if they plan to take or are taking any prescription medications, over-the-counter drugs or herbal products.
Pregnancy
- Remind patients to inform their healthcare provider immediately if they become pregnant or intend to become pregnant during treatment with Supexa OD tablet.
- Inform patients to not breast-feed if they are taking Supexa OD tablet.
12.0 Date of issue
06 January 2025
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Supexa OD is and what it is used for
- What you need to know before you take Supexa OD
- How to take Supexa OD
- Possible side effects
- How to store Supexa OD
- Contents of the pack and other information
1. What Supexa OD is and what it is used for
Supexa OD contains the active substance edoxaban and belongs to a group of medicines called anticoagulants. This medicine helps to prevent blood clots from forming. It works by blocking the activity of factor Xa, which is an important component of blood clotting.
Supexa OD is used in adults to:
- prevent blood clots in the brain (stroke) and other blood vessels in the body if you have a form of irregular heart rhythm called nonvalvular atrial fibrillation and at least one additional risk factor, such as heart failure, previous stroke or high blood pressure;
- treat blood clots in the veins of the legs (deep vein thrombosis) and in the blood vessels in the lungs (pulmonary embolism), and to prevent blood clots from re-occurring in the blood vessels in the legs and/or lungs.
2. What you need to know before you take Supexa OD
Do not take Supexa OD
- if you are allergic to edoxaban or any of the other ingredients of this medicine
- if you are actively bleeding
- if you have a disease or condition that increases the risk of serious bleeding (e.g. stomach ulcer, injury or bleeding in the brain, or recent surgery of the brain or eyes).
- if you are taking other medicines to prevent blood clotting (e.g. warfarin, dabigatran, rivaroxaban, apixaban or heparin), except when changing anticoagulant treatment or while getting heparin through a venous or arterial line to keep it open.
- if you have a liver disease which leads to an increased risk of bleeding.
- if you have uncontrolled high blood pressure.
- if you are pregnant or breast feeding.
Warnings and precautions
Talk to your doctor or pharmacist before taking Supexa OD,
if you have an increased risk of bleeding, as could be the case if you have any of the following conditions:
- end stage kidney disease or if you are on dialysis
- severe liver disease
- bleeding disorders
- a problem with the blood vessels in the back of your eyes (retinopathy)
- recent bleeding in your brain (intracranial or intracerebral bleeding)
- problems with the blood vessels in your brain or spinal column
If you have a mechanical heart valve.
Supexa OD 15 mg is only to be used when changing from Supexa OD 30 mg to a vitamin K antagonist (e.g. warfarin).
Take special care with Supexa OD,
- if you know that you have a disease called antiphospholipid syndrome (a disorder of the immune system that causes an increased risk for blood clots), tell your doctor who will decide if the treatment may need to be changed.
If you need to have an operation
- it is very important to take Supexa OD before and after the operation exactly at the times you have been told by your doctor. If possible, Supexa OD should be stopped at least 24 hours before an operation. Your doctor will determine when to restart Supexa OD. In emergency situations your physician will help determine the appropriate actions regarding Supexa OD.
Children and adolescents
Supexa OD is not recommended in children and adolescents under 18 years of age.
Other medicines and Supexa OD
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
If you are taking any of the following:
- some medicines for fungal infections (e.g. ketoconazole);
- medicines to treat abnormal heart beat (e.g. dronedarone, quinidine, verapamil);
- other medicines to reduce blood clotting (e.g. heparin, clopidogrel or vitamin K antagonists such as warfarin, acenocoumarol, phenprocoumon or dabigatran, rivaroxaban, apixaban);
- antibiotic medicines (e.g. erythromycin, clarithromycin);
- medicines to prevent organ rejection after transplantation (e.g. ciclosporin);
- anti-inflammatory and pain-relieving medicines (e.g. naproxen or acetylsalicylic acid);
- antidepressant medicines called selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors;
If any of the above apply to you, tell your doctor before taking Supexa OD, because these medicines may increase the effects of Supexa OD and the chance of unwanted bleeding. Your doctor will decide, if you should be treated with Supexa OD and if you should be kept under observation.
If you are taking any of the following:
- some medicines for treatment of epilepsy (e.g. phenytoin, carbamazepine, phenobarbital).
- St John’s Wort, a herbal product used for anxiety and mild depression.
- rifampicin, an antibiotic medicine.
If any of the above apply to you, tell your doctor before taking Supexa OD, because the effect of Supexa OD may be reduced. Your doctor will decide if you should be treated with Supexa OD and if you should be kept under observation.
Pregnancy and breast-feeding
Do not take Supexa OD if you are pregnant or breast-feeding. If there is a chance that you could become pregnant, use a reliable contraceptive while you are taking Supexa OD. If you become pregnant while you are taking Supexa OD, immediately tell your doctor, who will decide how you should be treated.
Driving and using machines
Supexa OD has no or negligible effects on your ability to drive or use machines.
3. How to take Supexa OD
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
How much to take?
The recommended dose is one 60 mg tablet once daily.
If you have impaired kidney function, the dose may be reduced to one 30 mg tablet once daily by your doctor.
if your body weight is 60 kg or lower, the recommended dose is one 30 mg tablet once daily;
if your doctor has prescribed medicines known as P-gp inhibitors: ciclosporin, dronedarone, erythromycin, or ketoconazole, the recommended dose is one 30 mg tablet once daily.
How to take the tablet?
Swallow the tablet, preferably with water.
Supexa OD can be taken with or without food.
If you have difficulty swallowing the tablet whole, talk to your doctor about other ways to take Supexa OD. The tablet may be crushed and mixed with water or apple puree immediately before you take it. If necessary, your doctor may also give you the crushed Supexa OD tablet through a tube via the nose (nasogastric tube) or a tube in the stomach (gastric feeding tube).
Your doctor may change your anticoagulant treatment as follows:
Changing from vitamin K antagonists (e.g. warfarin) to Supexa OD
Stop taking the vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to start taking Supexa OD.
Changing from non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban) to Supexa OD
Stop taking the previous medicines (e.g. dabigatran, rivaroxaban, or apixaban) and start Supexa OD at the time of the next scheduled dose.
Changing from parenteral anticoagulants (e.g. heparin) to Supexa OD
Stop taking the anticoagulant (e.g. heparin) and start Supexa OD at the time of the next scheduled anticoagulant dose.
Changing from Supexa OD to vitamin K antagonists (e.g. warfarin)
If you currently take 60 mg Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 30 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
If you currently take 30 mg (dose reduced) Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 15 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
Changing from Supexa OD to non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban)
Stop taking Supexa OD and start the non-VKA anticoagulant (e.g. dabigatran, rivaroxaban, or apixaban) at the time of the next scheduled dose of Supexa OD.
Changing from Supexa OD to parenteral anticoagulants (e.g. heparin)
Stop taking Supexa OD and start the parenteral anticoagulant (e.g. heparin) at the time of the next scheduled dose of Supexa OD.
Patients undergoing cardioversion:
If your abnormal heartbeat needs to be restored to normal by a procedure called cardioversion, take Supexa OD at the times your doctor tells you to prevent blood clots in the brain and other blood vessels in your body.
If you take more Supexa OD than you should
Tell your doctor immediately if you have taken too many Supexa OD tablets.
If you take more Supexa OD than recommended, you may have an increased risk of bleeding.
If you forget to take Supexa OD
You should take the tablet immediately and then continue the following day with the once daily tablet as usual. Do not take a double dose on the same day to make up for a forgotten dose.
If you stop taking Supexa OD
Do not stop taking Supexa OD without talking to your doctor first, because Supexa OD treats and prevents serious conditions.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them. Like other similar medicines (medicines to reduce blood clotting), Supexa OD may cause bleeding which may potentially be life-threatening. In some cases, the bleeding may not be obvious.
If you experience any bleeding event that does not stop by itself or if you experience signs of excessive bleeding (exceptional weakness, tiredness, paleness, dizziness, headache or unexplained swelling) consult your doctor immediately.
Your doctor may decide to keep you under closer observation or change your medicine.
Overall list of possible side effects:
Common (may affect up to 1 in 10 people)
- stomach ache
- abnormal liver blood tests
- bleeding from the skin or under the skin
- anaemia (low levels of red blood cells)
- bleeding from the nose
- bleeding from the vagina
- rash
- bleeding in the bowel
- bleeding from the mouth and/or throat
- blood found in your urine
- bleeding following an injury (puncture)
- bleeding in the stomach
- dizziness
- feeling sick
- headache
- itching
Uncommon (may affect up to 1 in 100 people)
- bleeding in the eyes
- bleeding from a surgical wound following an operation
- blood in the spit when coughing
- bleeding in the brain
- other types of bleeding
- reduced number of platelets in your blood (which can affect clotting)
- allergic reaction
- hives
Rare (may affect up to 1 in 1,000 people)
- bleeding in the muscles
- bleeding in joints
- bleeding in the abdomen
- bleeding in the heart
- bleeding inside the skull
- bleeding following a surgical procedure
- allergic shock
- swelling of any part of the body due to allergic reaction
Not known (frequency cannot be estimated from the available data)
- bleeding in the kidney sometimes with presence of blood in urine leading to inability of the kidneys to work properly (anticoagulant-related nephropathy).
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Supexa OD
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the carton and on each blister or bottle after EXP. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Supexa OD contains
- The active substance is edoxaban (as tosylate monohydrate).
Supexa OD 15 mg film-coated tablets
Each tablet contains 15 mg edoxaban (as tosylate monohydrate).
Supexa OD 30 mg film-coated tablets
Each tablet contains 30 mg edoxaban (as tosylate monohydrate).
Supexa OD 60 mg film-coated tablets
Each tablet contains 60 mg edoxaban (as tosylate monohydrate).
For More Information About This Product
Zuvilix Tablets 200mg
1.0 Generic name
Elagolix Tablets 150 mg / 200 mg
2.0 Qualitative and quantitative composition
Zuvilix 150 mg Each tablet contains :
155.2 mg of Elagolix Sodium
equivalent to Elagolix 150 mg
Colours : Titanium Dioxide IP, FD&C Red #40/, Allura Red AC, Aluminium Lake, FD&C Blue #1/, Brilliant Blue FCF, Aluminium Lake
Zuvilix 200 mg Each tablet contains :
207.0 mg of Elagolix Sodium
equivalent to Elagolix 200 mg
Colours : Titanium Dioxide IP, Ferric Oxide Red USP-NF
3.0 Dosage form and strength
Tablet 150 mg/200 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the management of moderate to severe pain associated with endometriosis.
4.2 Posology and method of administration
Limitations of use
Limit the duration of use based on the dose and coexisting condition.
Important dosing information
- Exclude pregnancy before starting Zuvilix or start Zuvilix within 7 days from the onset of menses.
- Take Zuvilix at approximately the same time each day, with or without food.
- Use the lowest effective dose, taking into account the severity of symptoms and treatment objectives.
- Limit the duration of use because of bone loss (Table 1).
Table 1. Recommended dosage and duration of use
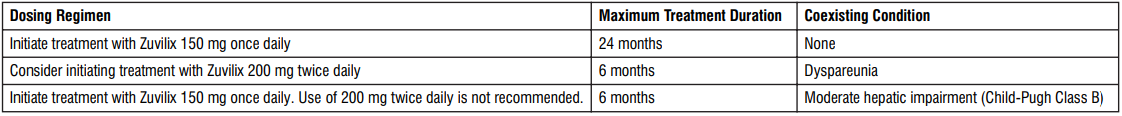
Renal impairment
No dose adjustment of Zuvilix is required in women with any degree of renal impairment or end-stage renal disease (including women on dialysis).
Hepatic impairment
No dosage adjustment of Zuvilix is required in women with mild hepatic impairment (Child-Pugh A). Compared to women with normal liver function, those with moderate hepatic impairment had approximately 3-fold higher elagolix exposures and those with severe hepatic impairment had approximately 7-fold higher elagolix exposures. Because of these increased exposures and risk for bone loss :
- Zuvilix 150 mg once daily is recommended for women with moderate hepatic impairment (Child-Pugh B) with the duration of treatment limited to 6 months. Use of Zuvilix 200 mg twice daily is not recommended for women with moderate hepatic impairment.
- Zuvilix is contraindicated in women with severe hepatic impairment (Child-Pugh C).
Missed dose
Instruct the patient to take a missed dose of Zuvilix on the same day as soon as she remembers and then resume the regular dosing schedule.
- 150 mg once daily : take no more than 1 tablet each day.
- 200 mg twice daily : take no more than 2 tablets each day.
4.3 Contraindications
- Pregnant. Exposure to elagolix early in pregnancy may increase the risk of early pregnancy loss.
- With known osteoporosis because of the risk of further bone loss.
- Severe hepatic impairment.
- Taking inhibitors of organic anion transporting polypeptide (OATP)1B1 (a hepatic uptake transporter) that are known or expected to significantly increase elagolix plasma concentrations.
- With known hypersensitivity reaction to elagolix or any of its inactive components. Reactions have included anaphylaxis and angioedema.
4.4 Special warnings and precautions for use
Bone loss
Elagolix causes a dose-dependent decrease in bone mineral density (BMD). BMD loss is greater with increasing duration of use and may not be completely reversible after stopping treatment. The impact of these BMD decreases on long-term bone health and future fracture risk are unknown. Elagolix is contraindicated in women with known osteoporosis. Consider assessment of BMD in patients with a history of a low-trauma fracture or other risk factors for osteoporosis or bone loss. Limit the duration of use to reduce the extent of bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients.
Change in menstrual bleeding pattern and reduced ability to recognize pregnancy
Women who take Elagolix may experience a reduction in the amount, intensity or duration of menstrual bleeding, which may reduce the ability to recognize the occurrence of a pregnancy in a timely manner. Perform pregnancy testing if pregnancy is suspected, and discontinue Elagolix if pregnancy is confirmed.
Suicidal ideation, suicidal behaviour, and exacerbation of mood disorders.
Suicidal ideation and behaviour, including one completed suicide, occurred in subjects treated with Elagolix in the endometriosis clinical trials. Elagolix subjects had a higher incidence of depression and mood changes compared to placebo, and Elagolix subjects with a history of suicidality or depression had a higher incidence of depression compared to subjects without such a history. Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benets. Patients with new or worsening depression, anxiety or other mood changes should be referred to a mental health professional, as appropriate. Advise patients to seek immediate medical attention for suicidal ideation and behaviour. Re-evaluate the benets and risks of continuing Elagolix if such events occur.
Hepatic transaminase elevations
In clinical trials, dose-dependent elevations of serum alanine aminotransferase (ALT) at least 3- times the upper limit of the reference range occurred with Elagolix. Use the lowest effective dose of Elagolix and instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice. Promptly evaluate patients with elevations in liver tests to determine whether the benefits of continued therapy outweigh the risks.
Interactions with hormonal contraceptives
Advise women to use effective non-hormonal contraceptives during treatment with Elagolix and for 28 days after discontinuing Elagolix. Increase in Estrogen Exposure and Potential Associated Increased Risks When Elagolix 200 mg Twice Daily is Taken with Combined Hormonal Contraceptives.
Potential for reduced efficacy of progestin-containing hormonal contraceptives
Co-administration of Elagolix 200 mg twice daily and a COC containing 0.1 mg levonorgestrel decreases the plasma concentrations of levonorgestrel by 27%, potentially affecting contraceptive efficacy. Co-administration of Elagolix with COCs containing norethindrone acetate did not show a reduction in plasma concentrations of norethindrone.
Reduced efficacy of Elagolix
Based on the mechanism of action of Elagolix, estrogen-containing contraceptives are expected to reduce the efficacy of Elagolix. The effect of progestin-only contraceptives on the efficacy of Elagolix is unknown.
4.5 Drugs interactions
Potential for Elagolix to affect other drugs
Elagolix is :
- A weak to moderate inducer of cytochrome P450 (CYP) 3A. Co-administration with Elagolix may decrease plasma concentrations of drugs that are substrates of CYP3A (see Table 2).
- A weak inhibitor of CYP 2C19. Co-administration with Elagolix may increase plasma concentrations of drugs that are substrates of CYP2C19 (see Table 2).
- An inhibitor of efux transporter P-glycoprotein (P-gp). Co-administration with Elagolix may increase plasma concentrations of drugs that are substrates of P-gp (see Table 2).
The effects of co-administration of Elagolix on concentrations of concomitant drugs and the clinical recommendations for these drug interactions are summarized in Table 2.
Table 2. Drug interactions : Effects of Elagolix on other drugs
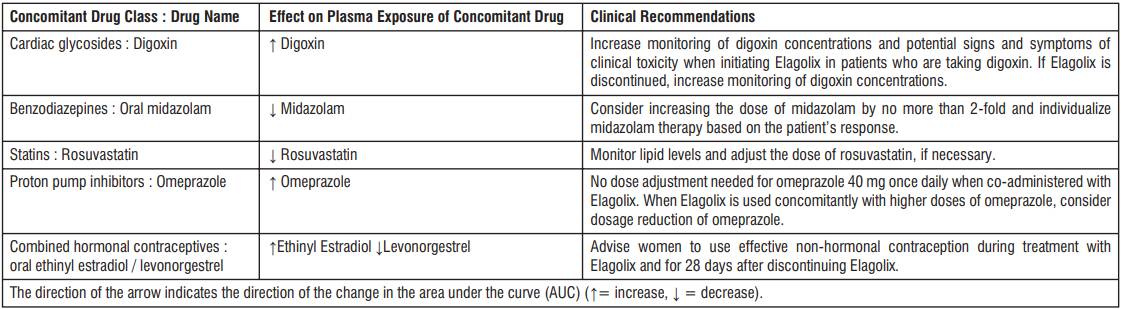
Potential for other drugs to affect Elagolix
Elagolix is a substrate of CYP3A, P-gp, and OATP1B1. Concomitant use of Elagolix 200 mg twice daily and strong CYP3A inhibitors for more than 1 month is not recommended. Limit concomitant use of Elagolix 150 mg once daily and strong CYP3A inhibitors to 6 months. Co-administration of Elagolix with strong CYP3A inducers may decrease Elagolix plasma concentrations and may result in a decrease of the therapeutic effects of Elagolix. Concomitant use of Elagolix 200 mg twice daily and rifampin is not recommended. Limit concomitant use of Elagolix 150 mg once daily and rifampin to 6 months. The effect of concomitant use of P-gp inhibitors or inducers on the pharmacokinetics of Elagolix is unknown. OATP1B1 inhibitors that are known or expected to signicantly increase Elagolix plasma concentrations are contraindicated due to increased risk of Elagolix associated adverse reactions.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Pregnancy
Use of Elagolix is contraindicated in pregnant women. Exposure to Elagolix early in pregnancy may increase the risk of early pregnancy loss. Discontinue Zuvilix if pregnancy occurs during treatment.
Lactation
There is no information on the presence of Elagolix or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. There are no adequate animal data on the excretion of Elagolix in milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Elagolix and any potential adverse effects on the breastfed child from Elagolix.
Females and males of reproductive potential
Based on the mechanism of action, there is a risk of early pregnancy loss if Elagolix is administered to a pregnant woman.
Pregnancy Testing
Elagolix may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding. Exclude pregnancy before initiating treatment with Elagolix. Perform pregnancy testing if pregnancy is suspected during treatment with Elagolix and discontinue treatment if pregnancy is confirmed.
Contraception
Advise women to use effective non-hormonal contraception during treatment with Elagolix and for 28 days after discontinuing Elagolix.
Pediatric use
Safety and effectiveness of Elagolix in pediatric patients have not been established.
4.7 Effects on ability to drive and use machines
Zuvilix is unlikely to affect the ability to drive and use machines as it is not distributed in the central nervous system.
4.8 Undesirable effects
The following serious adverse reactions are reported Bone loss, change in menstrual bleeding pattern and reduced ability to recognize pregnancy, suicidal ideation, suicidal behaviour, and exacerbation of mood disorders, hepatic transaminase elevations, hot flush, headache, nausea, insomnia, anxiety, mood altered, mood swings, amenorrhea, depressed mood, depression, depressive symptoms and/or tearfulness, appendicitis, abdominal pain, back pain, arthralgia, libido, diarrhoea, abdominal pain, weight gain, dizziness, constipation, irritability
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com
Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In case of overdose, monitor the patient for any signs or symptoms of adverse reactions and initiate appropriate symptomatic treatment, as needed.
5.0 Pharmacological properties
5.1 Mechanism of action
Elagolix is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of Elagolix results in dosedependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones, estradiol and progesterone.
5.2 Pharmacodynamic properties
Effect on Ovulation and Estradiol In a 3-menstrual cycle study in healthy women, Elagolix 150 mg once daily and 200 mg twice daily resulted in an ovulation rate of approximately 50% and 32%, respectively. In the Phase 3 trials in women with endometriosis, Elagolix caused a dose-dependent reduction in median estradiol concentrations to approximately 42 pg/mL for 150 mg once daily regimen and 12 pg/mL for the 200 mg twice daily regimen.
Cardiac Electrophysiology
The effect of Elagolix on the QTc interval was evaluated in a randomized, placebo- and positive-controlled, open-label, single-dose, crossover thorough QTc study in 48 healthy adult premenopausal women. Elagolix concentrations in subjects given a single dose of 1200 mg was 17-times higher than the concentration in subjects given Elagolix 200 mg twice daily. There was no clinically relevant prolongation of the QTc interval.
5.3 Pharmacokinetic properties
The pharmacokinetic properties of Elagolix in healthy subjects are summarized in Table 3. Table 3. Pharmacokinetic Properties of Elagolix in Healthy Subjects
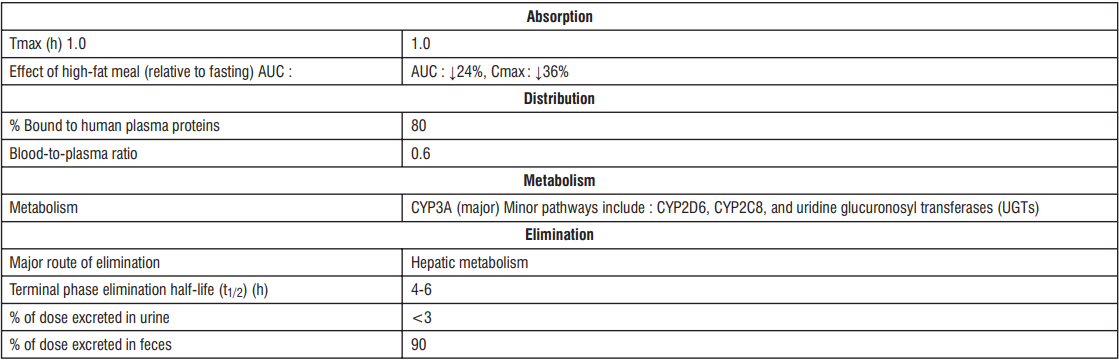
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Carcinogenesis, mutagenesis, impairment of fertility
Two-year carcinogenicity studies conducted in mice (50, 150, or 500 mg/kg/day) and rats (150, 300, or 800 mg/kg/day) that administered Elagolix by the dietary route revealed no increase in tumors in mice at up to 19-fold the MRHD based on AUC. In the rat, there was an increase in thyroid (male and female) and liver (males only) tumors at the high dose (12 to 13-fold the MRHD). The rat tumors were likely species-specic and of negligible relevance to humans. Elagolix was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, and the in vivo mouse micronucleus assay. In a fertility study conducted in the rat, there was no effect of Elagolix on fertility at any dose (50, 150, or 300 mg/kg/day). Based on AUC, the exposure multiple for the MRHD in women compared to the highest dose of 300 mg/kg/day in female rats is approximately 5-fold. However, because Elagolix has low afnity for the GnRH receptor in the rat and because effects on fertility are most likely to be mediated via the GnRH receptor, these data have low relevance to humans.
7.0 Description
Zuvilix tablets for oral administration contain elagolix sodium, the sodium salt of the active moiety elagolix. Elagolix sodium is a non-peptide small molecule GnRH receptor antagonist.
Elagolix sodium is chemically described as sodium 4-({(1R)-2-[5-(2-fluoro-3- methoxyphenyl)-3-{[2-fluoro-6-(trifluoromethyl)phenyl]methyl}-4-methyl-2,6-dioxo-3,6- dihydropyrimidin-1(2H)-yl]-1- phenylethyl}amino)butanoate.
Elagolix sodium has a molecular formula of C32H29F5N3O5Na and a molecular weight of 653.58
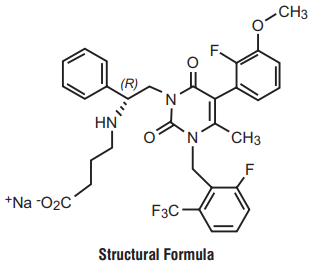
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
PVC-Alu blister strip of 10 tablets.
8.4 Storage and handling instructions
Do not store above 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient counseling information
Bone Loss
Inform patients about the risk of bone loss. Advise patients that supplementary calcium and vitamin D may be beneficial if dietary intake of calcium and vitamin D is not adequate.
Change in Menstrual Bleeding Pattern, Contraception and Pregnancy
Advise women that Zuvilix may delay the recognition of pregnancy because it may reduce the amount, intensity, or duration of menstrual bleeding. Advise patients to use effective non-hormonal contraception while taking Zuvilix and to discontinue Zuvilix if pregnancy is confirmed. Advise pregnant women that there is a pregnancy registry that monitors outcomes in women who become pregnant while treated with Zuvilix.
Suicidal ideation, suicidal behaviour, and exacerbation of mood disorders
Advise patients that suicidal ideation and exacerbation of mood disorders may occur with Zuvilix use. Instruct patients to seek immediate medical attention for suicidal ideation and behaviour and for new onset or worsening depression, anxiety, or other mood changes.
Liver injury
Advise patients to promptly seek medical attention in case of signs or symptoms that may reflect liver injury, such as jaundice.
Zuvilix missed dose instructions
Instruct a patient who miss a dose of Zuvilix to take the missed dose on the same day as soon as she remembers and then resume the regular dosing schedule :
- 150 mg once daily : no more than 1 tablet each day should be taken.
- 200 mg twice daily : no more than 2 tablets each day should be taken.
11.0 Details of permission or license number with date
5/MN/TS/2014/F/G dated 16 November 2024
12.0 Date of issue
16 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Zuvilix Tablet is and what it is used for
- What you need to know before you take Zuvilix Tablet
- How to take Zuvilix Tablet
- Possible side effects
- How to store Zuvilix Tablet
- Contents of the pack and other information
1. What Zuvilix Tablet is and what it is used for
Zuvilix has a medicine called Elagolix sodium. It helps reduce moderate to severe pain caused by endometriosis, a condition where tissue similar to the lining inside the uterus grows outside it.
2. What you need to know before you take Zuvilix Tablet
Do not take Zuvilix if you:
- You are pregnant or think you might be pregnant.
- You have osteoporosis (weak bones).
- You have severe liver problems.
- You are taking certain medicines that affect how your liver works.
- You are allergic to elagolix or any other ingredients in the medicine.
Warnings and precautions:
Bone Loss
- Zuvilix can make your bones weaker over time, and this might not fully get better after you stop taking it.
- The longer you use it; the more bone loss you might have.
- The long-term effects on bone health and fracture risk are not known.
- Do not use Zuvilix if you have osteoporosis (weak bones).
- Your doctor might check your bone health if you have a history of fractures or other risk factors.
- Limit how long you use Zuvilix to reduce bone loss.
- Taking calcium and vitamin D might help, but this hasn't been studied.
Changes in Menstrual Bleeding and Pregnancy Recognition
- Zuvilix can change your menstrual bleeding, making it lighter or shorter.
- This might make it harder to know if you are pregnant.
- If you think you might be pregnant, take a pregnancy test and stop Zuvilix if you are pregnant.
Suicidal Thoughts and Mood Changes
- Some people taking Zuvilix have had suicidal thoughts or behaviour, including one person who died by suicide.
- People taking Zuvilix had more depression and mood changes than those taking a placebo.
- If you have a history of depression or suicidal thoughts, you might be more likely to have these issues with Zuvilix.
- If you feel depressed or have mood changes, talk to your doctor to see if you should continue taking Zuvilix.
- Seek immediate medical help if you have suicidal thoughts or behaviour.
Liver Problems
- Zuvilix can cause increases in liver enzymes, which might indicate liver damage.
- Use the lowest effective dose and see a doctor if you have symptoms of liver problems, like jaundice (yellowing of the skin or eyes).
- Your doctor will check your liver tests to decide if you should keep taking Zuvilix.
Interactions with Hormonal Contraceptives
- Use non-hormonal birth control while taking Zuvilix and for 28 days after stopping it.
- Taking Zuvilix with combined hormonal contraceptives (like birth control pills) can increase estrogen levels and related risks.
- Zuvilix can reduce the effectiveness of some hormonal contraceptives.
- The effect of progestin-only contraceptives on Zuvilix is not known.
Pregnancy
Use of Elagolix is contraindicated in pregnant women. Exposure to Elagolix early in pregnancy may increase the risk of early pregnancy loss. Discontinue Zuvilix if pregnancy occurs during treatment.
Lactation
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Elagolix and any potential adverse effects on the breastfed child from Elagolix.
Females and males of reproductive potential
Based on the mechanism of action, there is a risk of early pregnancy loss if Elagolix is administered to a pregnant woman.
Pregnancy Testing
Elagolix may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding. Exclude pregnancy before initiating treatment with Elagolix. Perform pregnancy testing if pregnancy is suspected during treatment with Elagolix and discontinue treatment if pregnancy is confirmed.
Contraception
Advise women to use effective non-hormonal contraception during treatment with Elagolix and for 28 days after discontinuing Elagolix.
Pediatric use
Safety and effectiveness of Elagolix in pediatric patients have not been established.
Driving and using machines:
Zuvilix is unlikely to affect the ability to drive and use machines as it is not distributed in the central nervous system.
How Elagolix Affects Other Medications
Digoxin (for heart problems): Elagolix can increase digoxin levels. Your doctor should monitor digoxin levels and watch for signs of toxicity.
Midazolam (a sedative): Elagolix can lower midazolam levels. Your doctor might need to increase the dose of midazolam.
Rosuvastatin (for cholesterol): Elagolix can lower rosuvastatin levels. Your doctor should monitor your cholesterol and adjust the dose if needed.
Omeprazole (for acid reflux): Elagolix can increase omeprazole levels. No dose change is needed for 40 mg daily, but higher doses might need adjustment.
Hormonal Contraceptives (like birth control pills): Elagolix can increase estrogen and decrease progestin levels. Use non-hormonal birth control methods during and for 28 days after stopping Elagolix.
How Other Medications Affect Elagolix
Strong CYP3A Inhibitors (like some antibiotics and antifungals): These can increase Elagolix levels. Use Elagolix 150 mg once daily for up to 6 months. Do not use Elagolix 200 mg twice daily with these drugs.
Strong CYP3A Inducers (like some seizure medications): These can decrease Elagolix levels, making it less effective. Use Elagolix 150 mg once daily for up to 6 months. Do not use Elagolix 200 mg twice daily with these drugs.
OATP1B1 Inhibitors (like some cholesterol medications): These can significantly increase Elagolix levels. Do not use Elagolix with these drugs due to the risk of side effects.
General Recommendations
Non-Hormonal Contraceptives: Use non-hormonal birth control methods while taking Elagolix and for 28 days after stopping it.
Monitoring: Regularly check drug levels and how well the medications are working when taking Elagolix with other drugs.
3. How to take Zuvilix Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
How to Take Zuvilix
- Before Starting: Make sure you are not pregnant before you start taking Zuvilix. If you are starting Zuvilix, do so within 7 days from the start of your period.
- Daily Routine: Take Zuvilix at the same time every day. You can take it with or without food.
- Dosage: Use the lowest dose that works for you. Your doctor will decide the right dose based on your symptoms.
- Duration: Do not use Zuvilix for too long because it can cause bone loss. Follow your doctor's instructions on how long to take it.
Recommended Dosages
- 150 mg once daily: You can take this dose for up to 24 months.
- 200 mg twice daily: This dose is for severe pain during intercourse (dyspareunia) and can be taken for up to 6 months.
Moderate Liver Problems:
Women with moderate liver problems have higher levels of the drug in their body. They should take 150 mg once daily for up to 6 months. They should not take the 200 mg dose twice daily.
Kidney Problems (Renal Impairment)
No Change Needed: Women with any level of kidney problems, including those on dialysis, do not need to change their dose of Zuvilix.
Liver Problems (Hepatic Impairment)
Mild Liver Problems: Women with mild liver problems do not need to change their dose of Zuvilix.
Severe Liver Problems: Women with severe liver problems should not take Zuvilix at all because of the high levels of the drug and the risk of bone loss.
Missed Dose
If you miss a dose, take it as soon as you remember on the same day. Then continue with your regular schedule.
- 150 mg once daily: Do not take more than 1 tablet each day.
- 200 mg twice daily: Do not take more than 2 tablets each day.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
The following serious adverse reactions are reported
Bone loss, change in menstrual bleeding pattern and reduced ability to recognize pregnancy, suicidal ideation, suicidal behaviour, and exacerbation of mood disorders, hepatic transaminase, elevations, hot flush, headache, nausea, insomnia, anxiety, mood altered, mood swings, amenorrhea, depressed mood, depression, depressive symptoms and/or tearfulness, appendicitis, abdominal pain, back pain, arthralgia, libido, diarrhoea, abdominal pain, weight gain, dizziness, constipation, irritability
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly:
Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page.
Website link: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Zuvilix Tablet
Do not store above 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Zuvilix contains:
- The active ingredient is Elagolix Sodium.
- Other ingredients include Titanium Dioxide IP, FD&C Red #40/, Allura Red AC, Colours: Titanium Dioxide IP, Ferric Oxide Red USP-NF
What Zuvilix looks like and contents of the pack:
- Zuvilix comes in tablet form in strengths of 150 mg, and 200 mg.
- The tablets are packed in a PVC-Alu blister strip of 10 tablets.
This leaflet was last revised on: 12 March 2025
For More Information About This Product
Supexa- OD 15 Tablets
1.0 Generic name
Edoxaban tablet
2.0 Qualitative and quantitative composition
SUPEXA OD 15
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….15 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 30
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….30 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
SUPEXA OD 60
Each film-coated tablet contains
Edoxaban Tosylate Monohydrate
equivalent to Edoxaban ……….60 mg
Excipient…………………….q.s
Colours: Yellow Oxide of Iron and Titanium Dioxide IP
3.0 Dosage form and strength
Film coated tablet, 15 mg / 30 mg / 60 mg
4.0 Clinical particulars
4.1Therapeutic indications
- For prevention of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation (NVAF) with one or more risk factors, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke or transient ischaemic attack (TIA).
- For the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the prevention of recurrent DVT and PE in adults.
4.2 Posology and method of administration
Posology
Prevention of stroke and systemic embolism
- The recommended dose is 60 mg edoxaban once daily. Therapy with edoxaban in NVAF patients should be continued long term.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate or severe renal impairment (creatinine clearance (CrCl) 15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of the following P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- For elderly patients aged 80 years and above: dose reduced to 15 mg once daily.
Treatment of DVT, PE and prevention of recurrent DVT and PE (venous thromboembolism (VTE))
- The recommended dose is 60 mg edoxaban once daily following initial use of parenteral anticoagulant for at least 5 days. Edoxaban and initial parenteral anticoagulant should not be administered simultaneously. The duration of therapy for treatment of DVT and PE (VTE), and prevention of recurrent VTE should be individualized after careful assessment of the treatment benefit against the risk for bleeding. Short duration of therapy (at least 3 months) should be based on transient risk factors (e.g. recent surgery, trauma, immobilization) and longer durations should be based on permanent risk factors or idiopathic DVT or PE.
- The recommended dose is 30 mg edoxaban once daily in patients with one or more of the following clinical factors:
- Moderate to severe renal impairment (CrCl -15 - 50 mL/min)
- Low body weight ≤ 60 kg
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Prevention of venous thromboembolism (VTE) in patients undergoing orthopedic surgery for the lower limbs - Total knee or hip replacement, hip fracture surgery:
- The usual adult dosage is 30 mg of edoxaban administered orally once daily.
- Consider reducing the dose to 15 mg once daily if
- Renal impairment (CrCl 30 mL/min and ≤ 50 mL/min).
- Concomitant use of P-glycoprotein (P-gp) inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem.
- Edoxaban should be administered 12 hours after surgery and after confirming that there is no bleeding from the surgical wound, etc.
- Edoxaban should be at least 2 hours after the epidural catheter is removed or lumbar puncture is performed.
Table 1: Summary of Edoxaban Posology in NVAF and VTE (DVT and PE)
Missed dose
If a dose of edoxaban is missed, the dose should be taken immediately and then be continued the following day with the once-daily intake as recommended. The patient should not take double the prescribed dose on the same day to make up for a missed dose. Switching to and from edoxaban Continued anticoagulant therapy is important in patients with NVAF and VTE. There may be situations that warrant a change in anticoagulation therapy (Table 2).
Table 2: Switching of anticoagulant treatment in NVAF and VTE (DVT and PE)
Special populations
Elderly population
No dose reduction is required.
Renal impairment
Renal function should be assessed in all patients by calculating the CrCl prior to initiation of treatment with edoxaban to exclude patients with end stage renal disease (i.e. CrCl < 15 mL/min), to use the correct edoxaban dose in patients with CrCl 15 – 50 mL/min (30 mg once daily), in patients with CrCl > 50 mL/min (60 mg once daily) and when deciding on the use of edoxaban in patients with increased CrCl.
Renal function should also be assessed when a change in renal function is suspected during treatment (e.g. hypovolaemia, dehydration, and in case of concomitant use of certain medicinal products).
The method used to estimate renal function (CrCl in mL/min) during the clinical development of edoxaban was the Cockcroft-Gault method. The formula is as follows:
For creatinine in µ mol/L:
1.23× (140-age [years])× weight [kg] (× 0.85 if Female)
Serum creatinine [µmol/L]
For creatinine in mg/dL:
(140-age [years]) × weight [kg] (× 0.85 if Female)
72×Serum creatinine [mg/dL]
This method is recommended when assessing patients' CrCl prior to and during edoxaban treatment.
In patients with mild renal impairment (CrCl > 50 – 80 mL/min), the recommended dose is 60 mg edoxaban once daily.
In patients with moderate or severe renal impairment (CrCl 15 – 50 mL/min), the recommended dose is 30 mg edoxaban once daily.
In patients with end stage renal disease (ESRD) (CrCl < 15 mL/min) or on dialysis, the use of edoxaban is not recommended.
Hepatic impairment
Edoxaban is contraindicated in patients with hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
In patients with severe hepatic impairment edoxaban is not recommended.
In patients with mild to moderate hepatic impairment the recommended dose is 60 mg edoxaban once daily. Edoxaban should be used with caution in patients with mild to moderate hepatic impairment.
Patients with elevated liver enzymes (alanine aminotransferase (ALT) or aspartate transaminase (AST) > 2 x upper limit of normal (ULN)) or total bilirubin ≥ 1.5 x ULN, were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed.
Body weight
For patients with body weight ≤ 60 kg, the recommended dose is 30 mg edoxaban once daily).
Gender
No dose reduction is required.
Concomitant use of edoxaban with P-glycoprotein (P-gp) inhibitors
In patients concomitantly taking edoxaban and the following P-gp inhibitors: ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem, the recommended dose is 30 mg edoxaban once daily.
No dose reduction is required for concomitant use of amiodarone, quinidine or verapamil. The use of edoxaban with other P-gp inhibitors including HIV protease inhibitors has not been studied.
Patients undergoing cardioversion
Edoxaban can be initiated or continued in patients who may require cardioversion. For transoesophageal echocardiogram (TEE) guided cardioversion in patients not previously treated with anticoagulants, edoxaban treatment should be started at least 2 hours before cardioversion to ensure adequate anticoagulation. Cardioversion should be performed no later than 12 hours after the dose of edoxaban on the day of the procedure.
For all patients undergoing cardioversion: Confirmation should be sought prior to cardioversion that the patient has taken edoxaban as prescribed. Decisions on initiation and duration of treatment should follow established guidelines for anticoagulant treatment in patients undergoing cardioversion.
Paediatric population
Edoxaban is not recommended for use in children and adolescents from birth to 18 years of age with confirmed VTE (PE and/or DVT) event as the efficacy has not been established.
Method of administration
For oral use.
Edoxaban can be taken with or without food.
For patients who are unable to swallow whole tablets, edoxaban tablets may be crushed and mixed with water or apple puree and immediately administered orally. Alternatively, edoxaban tablets may be crushed and suspended in a small amount of water and immediately delivered through a nasogastric tube or gastric feeding tube after which it should be flushed with water. Crushed edoxaban tablets are stable in water and apple puree for up to 4 hours.
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in the formulation.
- Clinically significant active bleeding.
- Hepatic disease associated with coagulopathy and clinically relevant bleeding risk.
- Lesion or condition, if considered to be a significant risk for major bleeding. This may include current or recent gastrointestinal ulceration, presence of malignant neoplasms at high risk of bleeding, recent brain or spinal injury, recent brain, spinal or ophthalmic surgery, recent intracranial haemorrhage, known or suspected oesophageal varices, arteriovenous malformations, vascular aneurysms or major intraspinal or intracerebral vascular abnormalities.
- Uncontrolled severe hypertension.
- Concomitant treatment with any other anticoagulants e.g. UFH, LMWH (enoxaparin, dalteparin, etc.), heparin derivatives (fondaparinux, etc.), oral anticoagulants (warfarin, dabigatran etexilate, rivaroxaban, apixaban etc.) except under specific circumstances of switching oral anticoagulant therapy or when UFH is given at doses necessary to maintain an open central venous or arterial catheter.
- Pregnancy and breast-feeding.
4.4 Special warnings and precautions for use
Edoxaban 15 mg is not indicated as monotherapy, as it may result in decreased efficacy. It is only indicated in the process of switching from edoxaban 30 mg (patients with one or more clinical factors for increased exposure; see table 1) to VKA, together with an appropriate VKA dose.
Haemorrhagic risk
Edoxaban increases the risk of bleeding and can cause serious, potentially fatal bleeding. Edoxaban, like other anticoagulants, is recommended to be used with caution in patients with increased risk of bleeding. Edoxaban administration should be discontinued if severe haemorrhage occurs.
In the clinical studies mucosal bleedings (e.g. epistaxis, gastrointestinal, genitourinary) and anaemia were seen more frequently during long term edoxaban treatment compared with VKA treatment. Thus, in addition to adequate clinical surveillance, laboratory testing of haemoglobin/haematocrit could be of value to detect occult bleeding, as judged to be appropriate.
Several sub-groups of patients, as detailed below, are at increased risk of bleeding. These patients are to be carefully monitored for signs and symptoms of bleeding complications and anaemia after initiation of treatment. Any unexplained fall in haemoglobin or blood pressure should lead to a search for a bleeding site.
The anticoagulant effect of edoxaban cannot be reliably monitored with standard laboratory testing. A specific anticoagulant reversal agent for edoxaban is not available. Haemodialysis does not significantly contribute to edoxaban clearance.
Elderly
The co-administration of edoxaban with acetylsalicylic acid (ASA) in elderly patients should be used cautiously because of a potentially higher bleeding risk.
Renal impairment
The plasma area under the curve (AUC) for subjects with mild (CrCl > 50 - 80 mL/min), moderate (CrCl 30 - 50 mL/min) and severe (CrCl < 30 mL/min but not undergoing dialysis) renal impairment was increased by 32%, 74%, and 72%, respectively, relative to subjects with normal renal function. In patients with end stage renal disease or on dialysis, edoxaban is not recommended.
Renal function in NVAF A trend towards decreasing efficacy with increasing CrCl was observed for edoxaban compared to well-managed warfarin.
Edoxaban should be used in patients with NVAF and high CrCl only after a careful evaluation of the individual thromboembolic and bleeding risk.
Assessment of renal function: CrCl should be monitored at the beginning of the treatment in all patients and afterwards when clinically indicated.
Hepatic impairment
Edoxaban is not recommended in patients with severe hepatic impairment. Edoxaban should be used with caution in patients with mild or moderate hepatic impairment.
Patients with elevated liver enzymes (ALT/AST > 2 x ULN) or total bilirubin ≥ 1.5 x ULN were excluded in clinical studies. Therefore, edoxaban should be used with caution in this population. Prior to initiating edoxaban, liver function testing should be performed. Periodic hepatic monitoring is recommended for patients on edoxaban treatment beyond 1 year.
Discontinuation for surgery and other interventions
If anticoagulation must be discontinued to reduce the risk of bleeding with surgical or other procedures, edoxaban should be stopped as soon as possible and preferably at least 24 hours before the procedure.
In deciding whether a procedure should be delayed until 24 hours after the last dose of edoxaban, the increased risk of bleeding should be weighed against the urgency of the intervention. Edoxaban should be restarted after the surgical or other procedures as soon as adequate haemostasis has been established, noting that the time to onset of the edoxaban anticoagulant therapeutic effect is 1 – 2 hours. If oral medicinal products cannot be taken during or after surgical intervention, consider administering a parenteral anticoagulant and then switch to oral once daily edoxaban.
Interaction with other medicinal products affecting haemostasis
Concomitant use of medicinal products affecting haemostasis may increase the risk of bleeding. These include ASA, P2Y12 platelet inhibitors, other antithrombotic agents, fibrinolytic therapy, selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs), and chronic nonsteroidal anti-inflammatory drugs (NSAIDs).
Prosthetic heart valves and moderate to severe mitral stenosis
Edoxaban has not been studied in patients with mechanical heart valves, in patients during the first 3 months after implantation of a bioprosthetic heart valve, with or without atrial fibrillation, or in patients with moderate to severe mitral stenosis. Therefore, use of edoxaban is not recommended in these patients.
Haemodynamically unstable PE patients or patients who require thrombolysis or pulmonary embolectomy
Edoxaban is not recommended as an alternative to UFH in patients with pulmonary embolism who are haemodynamically unstable or may receive thrombolysis or pulmonary embolectomy since the safety and efficacy of edoxaban have not been established in these clinical situations.
Patients with active cancer
Efficacy and safety of edoxaban in the treatment and/or prevention of VTE in patients with active cancer have not been established.
Patients with antiphospholipid syndrome
Direct acting oral anticoagulants (DOACs) including edoxaban are not recommended for patients with a history of thrombosis who are diagnosed with antiphospholipid syndrome. In particular for patients that are triple positive (for lupus anticoagulant, anticardiolipin antibodies, and anti-beta 2-glycoprotein I antibodies), treatment with DOACs could be associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
Laboratory coagulation parameters
Although treatment with edoxaban does not require routine monitoring, the effect on anticoagulation can be estimated by a calibrated quantitative anti-Factor Xa (anti-FXa) assay which may help to inform clinical decisions in particular situations as, e.g. overdose and emergency surgery.
Edoxaban prolongs standard clotting tests such as prothrombin time (PT), INR, and activated partial thromboplastin time (aPTT) as a result of Factor Xa (FXa) inhibition. Changes observed in these clotting tests at the expected therapeutic dose are, however, small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of edoxaban.
4.5 Drug interactions
Edoxaban is predominantly absorbed in the upper gastrointestinal (GI) tract. Thus, medicinal products or disease conditions that increase gastric emptying and gut motility have the possibility of reducing edoxaban dissolution and absorption.
P-gp inhibitors
Edoxaban is a substrate for the efflux transporter P-gp. In pharmacokinetic (PK) studies, concomitant administration of edoxaban with the P-gp inhibitors ciclosporin, dronedarone, erythromycin, ketoconazole, quinidine, or verapamil resulted in increased plasma concentrations of edoxaban. Concomitant use of edoxaban with ciclosporin, dronedarone, erythromycin, ketoconazole, HIV protease inhibitors, azithromycin, clarithromycin, itraconazole, diltiazem requires dose reduction to 30 mg once daily. Concomitant use of edoxaban with quinidine, verapamil, or amiodarone does not require dose reduction based on clinical data.
The use of edoxaban with other P-gp inhibitors including human immunodeficiency virus (HIV) protease inhibitors has not been studied.
Edoxaban 30 mg instead of 60 mg once daily must be administered during concomitant use with the following P-gp inhibitors:
- Ciclosporin: Concurrent administration of a single dose of ciclosporin 500 mg with a single dose of edoxaban 60 mg increased edoxaban AUC and maximum serum concentration (Cmax) by 73% and 74%, respectively.
- Dronedarone: Dronedarone 400 mg twice daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 5 increased edoxaban AUC and Cmax by 85% and 46%, respectively.
- Erythromycin: Erythromycin 500 mg four times daily for 8 days with a single concomitant dose of edoxaban 60 mg on day 7 increased the edoxaban AUC and Cmax by 85% and 68%, respectively.
- Ketoconazole: Ketoconazole 400 mg once daily for 7 days with a single concomitant dose of edoxaban 60 mg on day 4, increased edoxaban AUC and Cmax by 87% and 89%, respectively.
Edoxaban 60 mg once daily is recommended during concomitant use with the following P-gp inhibitors:
- Quinidine: Quinidine 300 mg once daily on days 1 and 4 and three times daily on days 2 and 3, with a single concomitant dose of edoxaban 60 mg on day 3, increased edoxaban AUC over 24 hours by 77% and Cmax by 85%, respectively.
- Verapamil: Verapamil 240 mg once daily for 11 days with a single concomitant dose of edoxaban 60 mg on day 10 increased the edoxaban AUC and Cmax by approximately 53%.
- Amiodarone: Co-administration of amiodarone 400 mg once daily with edoxaban 60 mg once daily increased AUC by 40% and Cmax by 66%. This was not considered clinically significant. In ENGAGE AF-TIMI 48 study in NVAF, efficacy and safety results were similar for subjects with and without concomitant amiodarone use.
P-gp inducers
Co-administration of edoxaban with the P-gp inducer rifampicin led to a decrease in mean edoxaban AUC and a shortened half-life, with possible decreases in its pharmacodynamic effects. The concomitant use of edoxaban with other P-gp inducers (e.g. phenytoin, carbamazepine, phenobarbital or St. John's Wort) may lead to reduced edoxaban plasma concentrations. Edoxaban should be used with caution when co-administered with P-gp inducers.
P-gp substrates
Digoxin
Edoxaban 60 mg once daily on days 1 to 14 with co administration of multiple daily doses of digoxin 0.25 mg twice daily (days 8 and 9) and 0.25 mg once daily (days 10 to 14) increased the Cmax of edoxaban by 17%, with no significant effect on AUC or renal clearance at steady state. When the effects of edoxaban on digoxin PK were also examined, the Cmax of digoxin increased by approximately 28% and AUC by 7%. This was not considered clinically relevant. No dose modification is necessary when edoxaban is administered with digoxin.
Anticoagulants, antiplatelet, NSAIDs and SSRIs/SNRIs
Anticoagulants
Co-administration of edoxaban with other anticoagulants is contraindicated due to increased risk of bleeding.
ASA
Co-administration of ASA (100 mg or 325 mg) and edoxaban increased bleeding time relative to either medicinal product alone. Co-administration of high dose ASA (325 mg) increased the steady state Cmax and AUC of edoxaban by 35% and 32%, respectively. The concomitant chronic use of high dose ASA (325 mg) with edoxaban is not recommended. Concomitant administration of higher doses than 100 mg ASA should only be performed under medical supervision.
In clinical studies concomitant use of ASA (low dose ≤ 100 mg/day), other antiplatelet agents, and thienopyridines was permitted and resulted in approximately a 2-fold increase in major bleeding in comparison with no concomitant use, although to a similar extent in the edoxaban and warfarin groups. Co-administration of low dose ASA (≤ 100 mg) did not affect the peak or total exposure of edoxaban either after single dose or at steady-state. Edoxaban can be co-administered with low dose ASA (≤ 100 mg/day).
Platelet inhibitors
In ENGAGE AF-TIMI 48 concomitant use of thienopyridines (e.g. clopidogrel) monotherapy was permitted and resulted in increased clinically relevant bleeding although with a lower risk of bleeding on edoxaban compared to warfarin.
There is very limited experience on the use of edoxaban with dual antiplatelet therapy or fibrinolytic agents.
NSAIDs
Co-administration of naproxen and edoxaban increased bleeding time relative to either medicinal product alone. Naproxen had no effect on the Cmax and AUC of edoxaban. In clinical studies, co-administration of NSAIDs resulted in increased clinically relevant bleeding. Chronic use of NSAIDs with edoxaban is not recommended.
SSRIs/SNRIs
As with other anticoagulants the possibility may exist that patients are at increased risk of bleeding in case of concomitant use with SSRIs or SNRIs due to their reported effect on platelets.
Effect of edoxaban on other medicinal products
Edoxaban increased the Cmax of concomitantly administered digoxin by 28%; however, the AUC was not affected. Edoxaban had no effect on the Cmax and AUC of quinidine. Edoxaban decreased the Cmax and AUC of concomitantly administered verapamil by 14% and 16%, respectively.
4.6 Use in special populations (such as pregnant woman, lactating women, paediatric patients, geriatric patients etc.)
Women of childbearing potential
Women of childbearing potential should avoid becoming pregnant during treatment with edoxaban.
Pregnancy
Safety and efficacy of edoxaban have not been established in pregnant women. Studies in animals have shown reproductive toxicity. Due to the potential reproductive toxicity, the intrinsic risk of bleeding and the evidence that edoxaban passes the placenta, Supexa OD is contraindicated during pregnancy.
Breast-feeding
Safety and efficacy of edoxaban have not been established in breast-feeding women. Data from animals indicate that edoxaban is secreted into breast milk. Therefore, Supexa OD is contraindicated during breast-feeding. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from therapy.
Fertility
No specific studies with edoxaban in human beings have been conducted to evaluate effects on fertility. In a study on male and female fertility in rats no effects were seen
4.7 Effects on ability to drive and use machines
Edoxaban has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The most commonly reported adverse reactions associated with edoxaban treatment are epistaxis, haematuria and anaemia. Bleeding can occur at any site and may be severe and even fatal.
Table 3. Tabulated list of adverse reactions
Reporting of side effects
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via
email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Overdose
Overdose with Edoxaban may lead to haemorrhage. Experience with overdose cases is very limited. Studies have confirmed that Andexanet alfa, a modifed recombinant inactive form of human factor Xa that binds to and blocks the effects of factor Xa inhibitors can be used as an antidote1,2. Early administration of activated charcoal may be considered in case of Edoxaban overdose to reduce absorption. This recommendation is based on standard treatment of medicinal product overdose and data available with similar compounds, as the use of activated charcoal to reduce absorption of Edoxaban has not been specifically studied in the Edoxaban clinical programme.
Management of bleeding :
Should a bleeding complication arise in a patient receiving Edoxaban, the next Edoxaban administration should be delayed or treatment should be discontinued as appropriate. Edoxaban has a half-life of approximately 10 to 14 hours. Management should be individualized according to the severity and location of the haemorrhage. Appropriate symptomatic treatment could be used as needed, such as mechanical compression (e.g. for severe epistaxis), surgical haemostasis with bleeding control procedures, fluid replacement and haemodynamic support, blood products (packed red cells or fresh frozen plasma, depending on associated anaemia or coagulopathy) or platelets. For life-threatening bleeding that cannot be controlled with the measures such as transfusion or haemostasis, the administration of a 4-factor prothrombin complex concentrate (PCC) at 50 IU/kg has been shown to reverse the effects of Edoxaban 30 minutes after completing the infusion.
Recombinant factor VIIa (r-FVIIa) can also be considered. However, there is limited clinical experience with the use of this product in individuals receiving Edoxaban. Depending on local availability, a consultation with a coagulation expert should be considered in case of major bleedings. Protamine sulfate and vitamin K are not expected to affect the anticoagulant activity of Edoxaban. There is no experience with antifibrinolytic agents (Tranexamic acid, Aminocaproic acid) in individuals receiving Edoxaban. There is neither scientific rationale for benefit nor experience with the use of systemic haemostatics (Desmopressin, Aprotinin) in individuals receiving Edoxaban. Due to the high plasma protein binding Edoxaban is not expected to be dialyzable.
5.0 Pharmacological properties
5.1 Mechanism of action
Edoxaban is a highly selective, direct and reversible inhibitor of FXa, the serine protease located in the final common pathway of the coagulation cascade. Edoxaban inhibits free FXa, and prothrombinase activity. Inhibition of FXa in the coagulation cascade reduces thrombin generation, prolongs clotting time and reduces the risk of thrombus formation.
5.2 Pharmacodynamic properties
Edoxaban produces rapid onset of pharmacodynamic effects within 1 - 2 hours, which corresponds with peak Edoxaban exposure (Cmax). The pharmacodynamic effects measured by anti-FXa assay are predictable and correlate with the dose and the concentration of Edoxaban. As a result of FXa inhibition, Edoxaban also prolongs clotting time in tests such as PT and aPTT. Changes observed in these clotting tests are expected at the therapeutic dose, however, these changes are small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of Edoxaban.
Effects of coagulation markers when switching from Rivaroxaban, Dabigatran or Apixaban to Edoxaban : In clinical pharmacology studies, healthy subjects received Rivaroxaban 20 mg once daily, Dabigatran 150 mg twice daily or Apixaban 5 mg twice daily, followed by a single dose of Edoxaban 60 mg on day 4. The effect on PT and other coagulation biomarkers (e.g. anti-FXa, aPTT) was measured. Following the switch to Edoxaban on day 4 the PT was equivalent to day 3 of Rivaroxaban and Apixaban. For dabigatran higher aPTT activity was observed after Edoxaban administration with prior Dabigatran treatment compared to that after treatment with Edoxaban alone. This is considered to be due to the carry-over effect of dabigatran treatment, however, this did not lead to a prolongation of bleeding time.
Based on these data, when switching from these anticoagulants to Edoxaban, the first dose of Edoxaban can be initiated at the time of the next scheduled dose of the previous anticoagulant
5.3 Pharmacokinetic properties
The pharmacokinetic parameters of Edoxaban in plasma after a single dose of Supexa OD 60 were as follows.
Absorption : Edoxaban is absorbed with peak plasma concentrations within 1 - 2 hours following oral administration of Edoxaban tablets. The absolute bioavailability is approximately 62%. Food increases peak exposure of Edoxaban tablets to a varying extent, but has minimal effect on total exposure.Edoxaban is poorly soluble at pH of 6.0 or higher. Co-administration of proton-pump inhibitors had no relevant impact on Edoxaban exposure.
In a study with 30 healthy subjects, both mean AUC and Cmax values for 60 mg Edoxaban administered as a crushed tablet orally mixed in apple puree or via nasogastric tube suspended in water were bioequivalent to the intact tablet. Given the predictable, dose-proportional pharmacokinetic profile of Edoxaban, the bioavailability results from this study are likely applicable to lower Edoxaban doses.
Distribution : Disposition is biphasic. The volume of distribution is 107 (19.9) L mean (SD). In vitro plasma protein binding is approximately 55%. There is no clinically relevant accumulation of Edoxaban (accumulation ratio 1.14) with once daily dosing. Steady state concentrations are achieved within 3 days.
Biotransformation : Unchanged Edoxaban is the predominant form in plasma. Edoxaban is metabolized via hydrolysis (mediated by carboxylesterase 1), conjugation or oxidation by CYP3A4/5 (< 10%). Edoxaban has three active metabolites, the predominant metabolite (M-4), formed by hydrolysis, is active and reaches less than 10% of the exposure of the parent compound in healthy subjects. Exposure to the other metabolites is less than 5%. Edoxaban is a substrate for the efflux transporter P-gp, but not a substrate for uptake transporters such as organic anion transporter polypeptide OATP1B1, organic anion transporters OAT1 or OAT3 or organic cation transporter OCT2. Its active metabolite is a substrate for OATP1B1.
Elimination : In healthy subjects, the total clearance is reported as 22 (± 3) L/hour; 50% is renally cleared (11 L/hour). Renal clearance accounts for approximately 35% of the administered dose. Metabolism and biliary / intestinal excretion account for the remaining clearance.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Acute oral toxicity study in rat and mice : A daily oral administration of Edoxaban Tosylate Monohydrate was given for 14 consecutive days to twenty-five Wistar Rats and twenty-five Swiss Albino Mice (5 per group). Each group (G1, G2, G3, G4, and G5) was administered a dose of 0, 250, 500, 1000 and 2000 mg/kg bw respectively. No mortality was observed in any dose group throughout the experiment period. All animals appeared normal throughout the study period. No external or internal gross pathological changes were observed in any of the treated animals.
Repeated dose oral toxicity study in rabbit and rat : A daily single dose of Edoxaban Tosylate Monohydrate was given for 28 days to Rabbits (3/sex/group) and Wistar Rats (6/sex/group). The selected oral doses selected were 0 (Control), 1.55, 3.1 and 6.2 mg/kg bw for Rabbit and 0 (Control), 3.1, 6.2 and 12.4 mg/kg bw. In reversal groups, animals were received control and high-dose for 28 days and observed for the next 14 days. No mortality and morbidity were observed among all the groups of animals throughout the experiment period. No clinical signs were observed in any groups of the animals throughout the experiment period. No statistically significant difference in the body weight, body weight change and feed consumption of the main study group and reversal high-dose animals compared to the control group.
Reproductive toxicology : Edoxaban showed vaginal haemorrhage at higher doses in rats and rabbits but had no effects in the reproductive performance of parent rats. In rats, no effects on male or female fertility were seen.
In animal reproduction studies, rabbits showed increased incidence of gallbladder variations at a dose of 200 mg/kg which is approximately 65 times the maximum recommended human dose (MRHD) of 60 mg/day based on total body surface area in mg/m2. Increased post-implantation pregnancy losses occurred in rats at 300 mg/kg/day (approximately 49 times the MRHD) and in rabbits at 200 mg/kg/day (approximately 65 times the MRHD) respectively. Edoxaban was excreted in the breast milk of lactating rats. Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, or phototoxicity.
7.0 Description
Edoxaban, a factor Xa inhibitor, is supplied as Edoxaban tosylate monohydrate. The chemical name is N-(5-Chloropyridin-2-yl)- N′-[(1S,2R,4S)-4-(N, N-dimethylcarbamoyl)-2-(5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2- carboxamido)cyclohexyl] oxamide mono (4-methylbenzenesulfonate) monohydrate.
Molecular formula : C24H30ClN7O4S•C7H8O3S•H2O
Molecular weight : 738.27 g/mo
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Supexa OD 15 : Alu-Alu blister strip of 15 tablets.
Supexa OD 30 : Alu-Alu blister strip of 15 tablets.
Supexa OD 60 : Alu-Alu blister strip of 15 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- Advise patients to take Supexa OD tablet exactly as prescribed.
- Remind patients to not discontinue Supexa OD tablet without talking to the healthcare provider who prescribed it.
- Instruct patients to keep an adequate supply of tablets to ensure continuous dosing of Supexa OD tablet.
- Instruct patients who cannot swallow the tablet whole to crush Supexa OD tablet, combine with 2 to 3 ounces of water or applesauce and ingest immediately.
- Instruct patients who require a gastric tube to crush the Supexa OD tablet and mix it with 2 to 3 ounces of water before administering immediately via the gastric feeding tube.
- Inform patients that if a dose is missed, they should take Supexa OD tablet as soon as possible the same day and resume the normal dosing schedule the following day. The dose should not be doubled to make up for a missing dose.
Bleeding risk
- Advise patients that they may bleed more easily, may bleed longer or bruise more easily when treated with Supexa OD tablet.
- Instruct patients to report any unusual bleeding immediately to their healthcare provider.
- For patients that are having neuraxial anesthesia or spinal puncture, advise patients to watch for signs and symptoms of spinal or epidural hematoma, such as back pain, tingling, numbness (especially in the lower limbs), muscle weakness and stool or urine incontinence. If any of these symptoms occur, advise the patient to contact his or her physician immediately.
Invasive or surgical procedures
- Remind patients to inform their healthcare providers that they are taking Supexa OD tablet before any surgery, medical or dental procedure is scheduled.
Concomitant medication and herbals
- Remind patients to inform their healthcare providers and dentists if they plan to take or are taking any prescription medications, over-the-counter drugs or herbal products.
Pregnancy
- Remind patients to inform their healthcare provider immediately if they become pregnant or intend to become pregnant during treatment with Supexa OD tablet.
- Inform patients to not breast-feed if they are taking Supexa OD tablet.
12.0 Date of issue
06 January 2025
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Supexa OD is and what it is used for
- What you need to know before you take Supexa OD
- How to take Supexa OD
- Possible side effects
- How to store Supexa OD
- Contents of the pack and other information
1. What Supexa OD is and what it is used for
Supexa OD contains the active substance edoxaban and belongs to a group of medicines called anticoagulants. This medicine helps to prevent blood clots from forming. It works by blocking the activity of factor Xa, which is an important component of blood clotting.
Supexa OD is used in adults to:
- prevent blood clots in the brain (stroke) and other blood vessels in the body if you have a form of irregular heart rhythm called nonvalvular atrial fibrillation and at least one additional risk factor, such as heart failure, previous stroke or high blood pressure;
- treat blood clots in the veins of the legs (deep vein thrombosis) and in the blood vessels in the lungs (pulmonary embolism), and to prevent blood clots from re-occurring in the blood vessels in the legs and/or lungs.
2. What you need to know before you take Supexa OD
Do not take Supexa OD
- if you are allergic to edoxaban or any of the other ingredients of this medicine
- if you are actively bleeding
- if you have a disease or condition that increases the risk of serious bleeding (e.g. stomach ulcer, injury or bleeding in the brain, or recent surgery of the brain or eyes).
- if you are taking other medicines to prevent blood clotting (e.g. warfarin, dabigatran, rivaroxaban, apixaban or heparin), except when changing anticoagulant treatment or while getting heparin through a venous or arterial line to keep it open.
- if you have a liver disease which leads to an increased risk of bleeding.
- if you have uncontrolled high blood pressure.
- if you are pregnant or breast feeding.
Warnings and precautions
Talk to your doctor or pharmacist before taking Supexa OD,
if you have an increased risk of bleeding, as could be the case if you have any of the following conditions:
- end stage kidney disease or if you are on dialysis
- severe liver disease
- bleeding disorders
- a problem with the blood vessels in the back of your eyes (retinopathy)
- recent bleeding in your brain (intracranial or intracerebral bleeding)
- problems with the blood vessels in your brain or spinal column
If you have a mechanical heart valve.
Supexa OD 15 mg is only to be used when changing from Supexa OD 30 mg to a vitamin K antagonist (e.g. warfarin).
Take special care with Supexa OD,
- if you know that you have a disease called antiphospholipid syndrome (a disorder of the immune system that causes an increased risk for blood clots), tell your doctor who will decide if the treatment may need to be changed.
If you need to have an operation
- it is very important to take Supexa OD before and after the operation exactly at the times you have been told by your doctor. If possible, Supexa OD should be stopped at least 24 hours before an operation. Your doctor will determine when to restart Supexa OD. In emergency situations your physician will help determine the appropriate actions regarding Supexa OD.
Children and adolescents
Supexa OD is not recommended in children and adolescents under 18 years of age.
Other medicines and Supexa OD
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
If you are taking any of the following:
- some medicines for fungal infections (e.g. ketoconazole);
- medicines to treat abnormal heart beat (e.g. dronedarone, quinidine, verapamil);
- other medicines to reduce blood clotting (e.g. heparin, clopidogrel or vitamin K antagonists such as warfarin, acenocoumarol, phenprocoumon or dabigatran, rivaroxaban, apixaban);
- antibiotic medicines (e.g. erythromycin, clarithromycin);
- medicines to prevent organ rejection after transplantation (e.g. ciclosporin);
- anti-inflammatory and pain-relieving medicines (e.g. naproxen or acetylsalicylic acid);
- antidepressant medicines called selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors;
If any of the above apply to you, tell your doctor before taking Supexa OD, because these medicines may increase the effects of Supexa OD and the chance of unwanted bleeding. Your doctor will decide, if you should be treated with Supexa OD and if you should be kept under observation.
If you are taking any of the following:
- some medicines for treatment of epilepsy (e.g. phenytoin, carbamazepine, phenobarbital).
- St John’s Wort, a herbal product used for anxiety and mild depression.
- rifampicin, an antibiotic medicine.
If any of the above apply to you, tell your doctor before taking Supexa OD, because the effect of Supexa OD may be reduced. Your doctor will decide if you should be treated with Supexa OD and if you should be kept under observation.
Pregnancy and breast-feeding
Do not take Supexa OD if you are pregnant or breast-feeding. If there is a chance that you could become pregnant, use a reliable contraceptive while you are taking Supexa OD. If you become pregnant while you are taking Supexa OD, immediately tell your doctor, who will decide how you should be treated.
Driving and using machines
Supexa OD has no or negligible effects on your ability to drive or use machines.
3. How to take Supexa OD
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
How much to take?
The recommended dose is one 60 mg tablet once daily.
If you have impaired kidney function, the dose may be reduced to one 30 mg tablet once daily by your doctor.
if your body weight is 60 kg or lower, the recommended dose is one 30 mg tablet once daily;
if your doctor has prescribed medicines known as P-gp inhibitors: ciclosporin, dronedarone, erythromycin, or ketoconazole, the recommended dose is one 30 mg tablet once daily.
How to take the tablet?
Swallow the tablet, preferably with water.
Supexa OD can be taken with or without food.
If you have difficulty swallowing the tablet whole, talk to your doctor about other ways to take Supexa OD. The tablet may be crushed and mixed with water or apple puree immediately before you take it. If necessary, your doctor may also give you the crushed Supexa OD tablet through a tube via the nose (nasogastric tube) or a tube in the stomach (gastric feeding tube).
Your doctor may change your anticoagulant treatment as follows:
Changing from vitamin K antagonists (e.g. warfarin) to Supexa OD
Stop taking the vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to start taking Supexa OD.
Changing from non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban) to Supexa OD
Stop taking the previous medicines (e.g. dabigatran, rivaroxaban, or apixaban) and start Supexa OD at the time of the next scheduled dose.
Changing from parenteral anticoagulants (e.g. heparin) to Supexa OD
Stop taking the anticoagulant (e.g. heparin) and start Supexa OD at the time of the next scheduled anticoagulant dose.
Changing from Supexa OD to vitamin K antagonists (e.g. warfarin)
If you currently take 60 mg Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 30 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
If you currently take 30 mg (dose reduced) Supexa OD:
Your doctor will tell you to reduce your dose of Supexa OD to a 15 mg tablet once daily and to take it together with a vitamin K antagonist (e.g. warfarin). Your doctor will need to do blood measurements and will instruct you when to stop taking Supexa OD.
Changing from Supexa OD to non-VKA oral anticoagulants (dabigatran, rivaroxaban, or apixaban)
Stop taking Supexa OD and start the non-VKA anticoagulant (e.g. dabigatran, rivaroxaban, or apixaban) at the time of the next scheduled dose of Supexa OD.
Changing from Supexa OD to parenteral anticoagulants (e.g. heparin)
Stop taking Supexa OD and start the parenteral anticoagulant (e.g. heparin) at the time of the next scheduled dose of Supexa OD.
Patients undergoing cardioversion:
If your abnormal heartbeat needs to be restored to normal by a procedure called cardioversion, take Supexa OD at the times your doctor tells you to prevent blood clots in the brain and other blood vessels in your body.
If you take more Supexa OD than you should
Tell your doctor immediately if you have taken too many Supexa OD tablets.
If you take more Supexa OD than recommended, you may have an increased risk of bleeding.
If you forget to take Supexa OD
You should take the tablet immediately and then continue the following day with the once daily tablet as usual. Do not take a double dose on the same day to make up for a forgotten dose.
If you stop taking Supexa OD
Do not stop taking Supexa OD without talking to your doctor first, because Supexa OD treats and prevents serious conditions.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them. Like other similar medicines (medicines to reduce blood clotting), Supexa OD may cause bleeding which may potentially be life-threatening. In some cases, the bleeding may not be obvious.
If you experience any bleeding event that does not stop by itself or if you experience signs of excessive bleeding (exceptional weakness, tiredness, paleness, dizziness, headache or unexplained swelling) consult your doctor immediately.
Your doctor may decide to keep you under closer observation or change your medicine.
Overall list of possible side effects:
Common (may affect up to 1 in 10 people)
- stomach ache
- abnormal liver blood tests
- bleeding from the skin or under the skin
- anaemia (low levels of red blood cells)
- bleeding from the nose
- bleeding from the vagina
- rash
- bleeding in the bowel
- bleeding from the mouth and/or throat
- blood found in your urine
- bleeding following an injury (puncture)
- bleeding in the stomach
- dizziness
- feeling sick
- headache
- itching
Uncommon (may affect up to 1 in 100 people)
- bleeding in the eyes
- bleeding from a surgical wound following an operation
- blood in the spit when coughing
- bleeding in the brain
- other types of bleeding
- reduced number of platelets in your blood (which can affect clotting)
- allergic reaction
- hives
Rare (may affect up to 1 in 1,000 people)
- bleeding in the muscles
- bleeding in joints
- bleeding in the abdomen
- bleeding in the heart
- bleeding inside the skull
- bleeding following a surgical procedure
- allergic shock
- swelling of any part of the body due to allergic reaction
Not known (frequency cannot be estimated from the available data)
- bleeding in the kidney sometimes with presence of blood in urine leading to inability of the kidneys to work properly (anticoagulant-related nephropathy).
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page. Website link: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Supexa OD
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the carton and on each blister or bottle after EXP. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Supexa OD contains
- The active substance is edoxaban (as tosylate monohydrate).
Supexa OD 15 mg film-coated tablets
Each tablet contains 15 mg edoxaban (as tosylate monohydrate).
Supexa OD 30 mg film-coated tablets
Each tablet contains 30 mg edoxaban (as tosylate monohydrate).
Supexa OD 60 mg film-coated tablets
Each tablet contains 60 mg edoxaban (as tosylate monohydrate).
For More Information About This Product
Zuvilix Tablets 150mg
1.0 Generic name
Elagolix Tablets 150 mg / 200 mg
2.0 Qualitative and quantitative composition
Zuvilix 150 mg Each tablet contains :
155.2 mg of Elagolix Sodium
equivalent to Elagolix 150 mg
Colours : Titanium Dioxide IP, FD&C Red #40/, Allura Red AC, Aluminium Lake, FD&C Blue #1/, Brilliant Blue FCF, Aluminium Lake
Zuvilix 200 mg Each tablet contains :
207.0 mg of Elagolix Sodium
equivalent to Elagolix 200 mg
Colours : Titanium Dioxide IP, Ferric Oxide Red USP-NF
3.0 Dosage form and strength
Tablet 150 mg/200 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the management of moderate to severe pain associated with endometriosis.
4.2 Posology and method of administration
Limitations of use
Limit the duration of use based on the dose and coexisting condition.
Important dosing information
- Exclude pregnancy before starting Zuvilix or start Zuvilix within 7 days from the onset of menses.
- Take Zuvilix at approximately the same time each day, with or without food.
- Use the lowest effective dose, taking into account the severity of symptoms and treatment objectives.
- Limit the duration of use because of bone loss (Table 1).
Table 1. Recommended dosage and duration of use
Renal impairment
No dose adjustment of Zuvilix is required in women with any degree of renal impairment or end-stage renal disease (including women on dialysis).
Hepatic impairment
No dosage adjustment of Zuvilix is required in women with mild hepatic impairment (Child-Pugh A). Compared to women with normal liver function, those with moderate hepatic impairment had approximately 3-fold higher elagolix exposures and those with severe hepatic impairment had approximately 7-fold higher elagolix exposures. Because of these increased exposures and risk for bone loss :
- Zuvilix 150 mg once daily is recommended for women with moderate hepatic impairment (Child-Pugh B) with the duration of treatment limited to 6 months. Use of Zuvilix 200 mg twice daily is not recommended for women with moderate hepatic impairment.
- Zuvilix is contraindicated in women with severe hepatic impairment (Child-Pugh C).
Missed dose
Instruct the patient to take a missed dose of Zuvilix on the same day as soon as she remembers and then resume the regular dosing schedule.
- 150 mg once daily : take no more than 1 tablet each day.
- 200 mg twice daily : take no more than 2 tablets each day.
4.3 Contraindications
- Pregnant. Exposure to elagolix early in pregnancy may increase the risk of early pregnancy loss.
- With known osteoporosis because of the risk of further bone loss.
- Severe hepatic impairment.
- Taking inhibitors of organic anion transporting polypeptide (OATP)1B1 (a hepatic uptake transporter) that are known or expected to significantly increase elagolix plasma concentrations.
- With known hypersensitivity reaction to elagolix or any of its inactive components. Reactions have included anaphylaxis and angioedema.
4.4 Special warnings and precautions for use
Bone loss
Elagolix causes a dose-dependent decrease in bone mineral density (BMD). BMD loss is greater with increasing duration of use and may not be completely reversible after stopping treatment. The impact of these BMD decreases on long-term bone health and future fracture risk are unknown. Elagolix is contraindicated in women with known osteoporosis. Consider assessment of BMD in patients with a history of a low-trauma fracture or other risk factors for osteoporosis or bone loss. Limit the duration of use to reduce the extent of bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients.
Change in menstrual bleeding pattern and reduced ability to recognize pregnancy
Women who take Elagolix may experience a reduction in the amount, intensity or duration of menstrual bleeding, which may reduce the ability to recognize the occurrence of a pregnancy in a timely manner. Perform pregnancy testing if pregnancy is suspected, and discontinue Elagolix if pregnancy is confirmed.
Suicidal ideation, suicidal behaviour, and exacerbation of mood disorders.
Suicidal ideation and behaviour, including one completed suicide, occurred in subjects treated with Elagolix in the endometriosis clinical trials. Elagolix subjects had a higher incidence of depression and mood changes compared to placebo, and Elagolix subjects with a history of suicidality or depression had a higher incidence of depression compared to subjects without such a history. Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benets. Patients with new or worsening depression, anxiety or other mood changes should be referred to a mental health professional, as appropriate. Advise patients to seek immediate medical attention for suicidal ideation and behaviour. Re-evaluate the benets and risks of continuing Elagolix if such events occur.
Hepatic transaminase elevations
In clinical trials, dose-dependent elevations of serum alanine aminotransferase (ALT) at least 3- times the upper limit of the reference range occurred with Elagolix. Use the lowest effective dose of Elagolix and instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice. Promptly evaluate patients with elevations in liver tests to determine whether the benefits of continued therapy outweigh the risks.
Interactions with hormonal contraceptives
Advise women to use effective non-hormonal contraceptives during treatment with Elagolix and for 28 days after discontinuing Elagolix. Increase in Estrogen Exposure and Potential Associated Increased Risks When Elagolix 200 mg Twice Daily is Taken with Combined Hormonal Contraceptives.
Potential for reduced efficacy of progestin-containing hormonal contraceptives
Co-administration of Elagolix 200 mg twice daily and a COC containing 0.1 mg levonorgestrel decreases the plasma concentrations of levonorgestrel by 27%, potentially affecting contraceptive efficacy. Co-administration of Elagolix with COCs containing norethindrone acetate did not show a reduction in plasma concentrations of norethindrone.
Reduced efficacy of Elagolix
Based on the mechanism of action of Elagolix, estrogen-containing contraceptives are expected to reduce the efficacy of Elagolix. The effect of progestin-only contraceptives on the efficacy of Elagolix is unknown.
4.5 Drugs interactions
Potential for Elagolix to affect other drugs
Elagolix is :
- A weak to moderate inducer of cytochrome P450 (CYP) 3A. Co-administration with Elagolix may decrease plasma concentrations of drugs that are substrates of CYP3A (see Table 2).
- A weak inhibitor of CYP 2C19. Co-administration with Elagolix may increase plasma concentrations of drugs that are substrates of CYP2C19 (see Table 2).
- An inhibitor of efux transporter P-glycoprotein (P-gp). Co-administration with Elagolix may increase plasma concentrations of drugs that are substrates of P-gp (see Table 2).
The effects of co-administration of Elagolix on concentrations of concomitant drugs and the clinical recommendations for these drug interactions are summarized in Table 2.
Table 2. Drug interactions : Effects of Elagolix on other drugs
Potential for other drugs to affect Elagolix
Elagolix is a substrate of CYP3A, P-gp, and OATP1B1. Concomitant use of Elagolix 200 mg twice daily and strong CYP3A inhibitors for more than 1 month is not recommended. Limit concomitant use of Elagolix 150 mg once daily and strong CYP3A inhibitors to 6 months. Co-administration of Elagolix with strong CYP3A inducers may decrease Elagolix plasma concentrations and may result in a decrease of the therapeutic effects of Elagolix. Concomitant use of Elagolix 200 mg twice daily and rifampin is not recommended. Limit concomitant use of Elagolix 150 mg once daily and rifampin to 6 months. The effect of concomitant use of P-gp inhibitors or inducers on the pharmacokinetics of Elagolix is unknown. OATP1B1 inhibitors that are known or expected to signicantly increase Elagolix plasma concentrations are contraindicated due to increased risk of Elagolix associated adverse reactions.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Pregnancy
Use of Elagolix is contraindicated in pregnant women. Exposure to Elagolix early in pregnancy may increase the risk of early pregnancy loss. Discontinue Zuvilix if pregnancy occurs during treatment.
Lactation
There is no information on the presence of Elagolix or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. There are no adequate animal data on the excretion of Elagolix in milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Elagolix and any potential adverse effects on the breastfed child from Elagolix.
Females and males of reproductive potential
Based on the mechanism of action, there is a risk of early pregnancy loss if Elagolix is administered to a pregnant woman.
Pregnancy Testing
Elagolix may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding. Exclude pregnancy before initiating treatment with Elagolix. Perform pregnancy testing if pregnancy is suspected during treatment with Elagolix and discontinue treatment if pregnancy is confirmed.
Contraception
Advise women to use effective non-hormonal contraception during treatment with Elagolix and for 28 days after discontinuing Elagolix.
Pediatric use
Safety and effectiveness of Elagolix in pediatric patients have not been established.
4.7 Effects on ability to drive and use machines
Zuvilix is unlikely to affect the ability to drive and use machines as it is not distributed in the central nervous system.
4.8 Undesirable effects
The following serious adverse reactions are reported Bone loss, change in menstrual bleeding pattern and reduced ability to recognize pregnancy, suicidal ideation, suicidal behaviour, and exacerbation of mood disorders, hepatic transaminase elevations, hot flush, headache, nausea, insomnia, anxiety, mood altered, mood swings, amenorrhea, depressed mood, depression, depressive symptoms and/or tearfulness, appendicitis, abdominal pain, back pain, arthralgia, libido, diarrhoea, abdominal pain, weight gain, dizziness, constipation, irritability
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com
Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In case of overdose, monitor the patient for any signs or symptoms of adverse reactions and initiate appropriate symptomatic treatment, as needed.
5.0 Pharmacological properties
5.1 Mechanism of action
Elagolix is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of Elagolix results in dosedependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones, estradiol and progesterone.
5.2 Pharmacodynamic properties
Effect on Ovulation and Estradiol In a 3-menstrual cycle study in healthy women, Elagolix 150 mg once daily and 200 mg twice daily resulted in an ovulation rate of approximately 50% and 32%, respectively. In the Phase 3 trials in women with endometriosis, Elagolix caused a dose-dependent reduction in median estradiol concentrations to approximately 42 pg/mL for 150 mg once daily regimen and 12 pg/mL for the 200 mg twice daily regimen.
Cardiac Electrophysiology
The effect of Elagolix on the QTc interval was evaluated in a randomized, placebo- and positive-controlled, open-label, single-dose, crossover thorough QTc study in 48 healthy adult premenopausal women. Elagolix concentrations in subjects given a single dose of 1200 mg was 17-times higher than the concentration in subjects given Elagolix 200 mg twice daily. There was no clinically relevant prolongation of the QTc interval.
5.3 Pharmacokinetic properties
The pharmacokinetic properties of Elagolix in healthy subjects are summarized in Table 3. Table 3. Pharmacokinetic Properties of Elagolix in Healthy Subjects
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Carcinogenesis, mutagenesis, impairment of fertility
Two-year carcinogenicity studies conducted in mice (50, 150, or 500 mg/kg/day) and rats (150, 300, or 800 mg/kg/day) that administered Elagolix by the dietary route revealed no increase in tumors in mice at up to 19-fold the MRHD based on AUC. In the rat, there was an increase in thyroid (male and female) and liver (males only) tumors at the high dose (12 to 13-fold the MRHD). The rat tumors were likely species-specic and of negligible relevance to humans. Elagolix was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, and the in vivo mouse micronucleus assay. In a fertility study conducted in the rat, there was no effect of Elagolix on fertility at any dose (50, 150, or 300 mg/kg/day). Based on AUC, the exposure multiple for the MRHD in women compared to the highest dose of 300 mg/kg/day in female rats is approximately 5-fold. However, because Elagolix has low afnity for the GnRH receptor in the rat and because effects on fertility are most likely to be mediated via the GnRH receptor, these data have low relevance to humans.
7.0 Description
Zuvilix tablets for oral administration contain elagolix sodium, the sodium salt of the active moiety elagolix. Elagolix sodium is a non-peptide small molecule GnRH receptor antagonist.
Elagolix sodium is chemically described as sodium 4-({(1R)-2-[5-(2-fluoro-3- methoxyphenyl)-3-{[2-fluoro-6-(trifluoromethyl)phenyl]methyl}-4-methyl-2,6-dioxo-3,6- dihydropyrimidin-1(2H)-yl]-1- phenylethyl}amino)butanoate.
Elagolix sodium has a molecular formula of C32H29F5N3O5Na and a molecular weight of 653.58
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
PVC-Alu blister strip of 10 tablets.
8.4 Storage and handling instructions
Do not store above 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient counseling information
Bone Loss
Inform patients about the risk of bone loss. Advise patients that supplementary calcium and vitamin D may be beneficial if dietary intake of calcium and vitamin D is not adequate.
Change in Menstrual Bleeding Pattern, Contraception and Pregnancy
Advise women that Zuvilix may delay the recognition of pregnancy because it may reduce the amount, intensity, or duration of menstrual bleeding. Advise patients to use effective non-hormonal contraception while taking Zuvilix and to discontinue Zuvilix if pregnancy is confirmed. Advise pregnant women that there is a pregnancy registry that monitors outcomes in women who become pregnant while treated with Zuvilix.
Suicidal ideation, suicidal behaviour, and exacerbation of mood disorders
Advise patients that suicidal ideation and exacerbation of mood disorders may occur with Zuvilix use. Instruct patients to seek immediate medical attention for suicidal ideation and behaviour and for new onset or worsening depression, anxiety, or other mood changes.
Liver injury
Advise patients to promptly seek medical attention in case of signs or symptoms that may reflect liver injury, such as jaundice.
Zuvilix missed dose instructions
Instruct a patient who miss a dose of Zuvilix to take the missed dose on the same day as soon as she remembers and then resume the regular dosing schedule :
- 150 mg once daily : no more than 1 tablet each day should be taken.
- 200 mg twice daily : no more than 2 tablets each day should be taken.
12.0 Date of issue
16 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Zuvilix Tablet is and what it is used for
- What you need to know before you take Zuvilix Tablet
- How to take Zuvilix Tablet
- Possible side effects
- How to store Zuvilix Tablet
- Contents of the pack and other information
1. What Zuvilix Tablet is and what it is used for
Zuvilix has a medicine called Elagolix sodium. It helps reduce moderate to severe pain caused by endometriosis, a condition where tissue similar to the lining inside the uterus grows outside it.
2. What you need to know before you take Zuvilix Tablet
Do not take Zuvilix if you:
- You are pregnant or think you might be pregnant.
- You have osteoporosis (weak bones).
- You have severe liver problems.
- You are taking certain medicines that affect how your liver works.
- You are allergic to elagolix or any other ingredients in the medicine.
Warnings and precautions:
Bone Loss
- Zuvilix can make your bones weaker over time, and this might not fully get better after you stop taking it.
- The longer you use it; the more bone loss you might have.
- The long-term effects on bone health and fracture risk are not known.
- Do not use Zuvilix if you have osteoporosis (weak bones).
- Your doctor might check your bone health if you have a history of fractures or other risk factors.
- Limit how long you use Zuvilix to reduce bone loss.
- Taking calcium and vitamin D might help, but this hasn't been studied.
Changes in Menstrual Bleeding and Pregnancy Recognition
- Zuvilix can change your menstrual bleeding, making it lighter or shorter.
- This might make it harder to know if you are pregnant.
- If you think you might be pregnant, take a pregnancy test and stop Zuvilix if you are pregnant.
Suicidal Thoughts and Mood Changes
- Some people taking Zuvilix have had suicidal thoughts or behaviour, including one person who died by suicide.
- People taking Zuvilix had more depression and mood changes than those taking a placebo.
- If you have a history of depression or suicidal thoughts, you might be more likely to have these issues with Zuvilix.
- If you feel depressed or have mood changes, talk to your doctor to see if you should continue taking Zuvilix.
- Seek immediate medical help if you have suicidal thoughts or behaviour.
Liver Problems
- Zuvilix can cause increases in liver enzymes, which might indicate liver damage.
- Use the lowest effective dose and see a doctor if you have symptoms of liver problems, like jaundice (yellowing of the skin or eyes).
- Your doctor will check your liver tests to decide if you should keep taking Zuvilix.
Interactions with Hormonal Contraceptives
- Use non-hormonal birth control while taking Zuvilix and for 28 days after stopping it.
- Taking Zuvilix with combined hormonal contraceptives (like birth control pills) can increase estrogen levels and related risks.
- Zuvilix can reduce the effectiveness of some hormonal contraceptives.
- The effect of progestin-only contraceptives on Zuvilix is not known.
Pregnancy
Use of Elagolix is contraindicated in pregnant women. Exposure to Elagolix early in pregnancy may increase the risk of early pregnancy loss. Discontinue Zuvilix if pregnancy occurs during treatment.
Lactation
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Elagolix and any potential adverse effects on the breastfed child from Elagolix.
Females and males of reproductive potential
Based on the mechanism of action, there is a risk of early pregnancy loss if Elagolix is administered to a pregnant woman.
Pregnancy Testing
Elagolix may delay the ability to recognize the occurrence of a pregnancy because it may reduce the intensity, duration, and amount of menstrual bleeding. Exclude pregnancy before initiating treatment with Elagolix. Perform pregnancy testing if pregnancy is suspected during treatment with Elagolix and discontinue treatment if pregnancy is confirmed.
Contraception
Advise women to use effective non-hormonal contraception during treatment with Elagolix and for 28 days after discontinuing Elagolix.
Pediatric use
Safety and effectiveness of Elagolix in pediatric patients have not been established.
Driving and using machines:
Zuvilix is unlikely to affect the ability to drive and use machines as it is not distributed in the central nervous system.
How Elagolix Affects Other Medications
Digoxin (for heart problems): Elagolix can increase digoxin levels. Your doctor should monitor digoxin levels and watch for signs of toxicity.
Midazolam (a sedative): Elagolix can lower midazolam levels. Your doctor might need to increase the dose of midazolam.
Rosuvastatin (for cholesterol): Elagolix can lower rosuvastatin levels. Your doctor should monitor your cholesterol and adjust the dose if needed.
Omeprazole (for acid reflux): Elagolix can increase omeprazole levels. No dose change is needed for 40 mg daily, but higher doses might need adjustment.
Hormonal Contraceptives (like birth control pills): Elagolix can increase estrogen and decrease progestin levels. Use non-hormonal birth control methods during and for 28 days after stopping Elagolix.
How Other Medications Affect Elagolix
Strong CYP3A Inhibitors (like some antibiotics and antifungals): These can increase Elagolix levels. Use Elagolix 150 mg once daily for up to 6 months. Do not use Elagolix 200 mg twice daily with these drugs.
Strong CYP3A Inducers (like some seizure medications): These can decrease Elagolix levels, making it less effective. Use Elagolix 150 mg once daily for up to 6 months. Do not use Elagolix 200 mg twice daily with these drugs.
OATP1B1 Inhibitors (like some cholesterol medications): These can significantly increase Elagolix levels. Do not use Elagolix with these drugs due to the risk of side effects.
General Recommendations
Non-Hormonal Contraceptives: Use non-hormonal birth control methods while taking Elagolix and for 28 days after stopping it.
Monitoring: Regularly check drug levels and how well the medications are working when taking Elagolix with other drugs.
3. How to take Zuvilix Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
How to Take Zuvilix
- Before Starting: Make sure you are not pregnant before you start taking Zuvilix. If you are starting Zuvilix, do so within 7 days from the start of your period.
- Daily Routine: Take Zuvilix at the same time every day. You can take it with or without food.
- Dosage: Use the lowest dose that works for you. Your doctor will decide the right dose based on your symptoms.
- Duration: Do not use Zuvilix for too long because it can cause bone loss. Follow your doctor's instructions on how long to take it.
Recommended Dosages
- 150 mg once daily: You can take this dose for up to 24 months.
- 200 mg twice daily: This dose is for severe pain during intercourse (dyspareunia) and can be taken for up to 6 months.
Moderate Liver Problems:
Women with moderate liver problems have higher levels of the drug in their body. They should take 150 mg once daily for up to 6 months. They should not take the 200 mg dose twice daily.
Kidney Problems (Renal Impairment)
No Change Needed: Women with any level of kidney problems, including those on dialysis, do not need to change their dose of Zuvilix.
Liver Problems (Hepatic Impairment)
Mild Liver Problems: Women with mild liver problems do not need to change their dose of Zuvilix.
Severe Liver Problems: Women with severe liver problems should not take Zuvilix at all because of the high levels of the drug and the risk of bone loss.
Missed Dose
If you miss a dose, take it as soon as you remember on the same day. Then continue with your regular schedule.
- 150 mg once daily: Do not take more than 1 tablet each day.
- 200 mg twice daily: Do not take more than 2 tablets each day.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
The following serious adverse reactions are reported
Bone loss, change in menstrual bleeding pattern and reduced ability to recognize pregnancy, suicidal ideation, suicidal behaviour, and exacerbation of mood disorders, hepatic transaminase, elevations, hot flush, headache, nausea, insomnia, anxiety, mood altered, mood swings, amenorrhea, depressed mood, depression, depressive symptoms and/or tearfulness, appendicitis, abdominal pain, back pain, arthralgia, libido, diarrhoea, abdominal pain, weight gain, dizziness, constipation, irritability
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly:
Website: www.zuventus.com and click the tab “Safety Reporting” located on the top end of the home page.
Website link: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Zuvilix Tablet
Do not store above 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Zuvilix contains:
- The active ingredient is Elagolix Sodium.
- Other ingredients include Titanium Dioxide IP, FD&C Red #40/, Allura Red AC, Colours: Titanium Dioxide IP, Ferric Oxide Red USP-NF
What Zuvilix looks like and contents of the pack:
- Zuvilix comes in tablet form in strengths of 150 mg, and 200 mg.
- The tablets are packed in a PVC-Alu blister strip of 10 tablets.
This leaflet was last revised on: 12 March 2025
For More Information About This Product
Trelagliptin Tablets 100 mg
1.0 Generic Name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
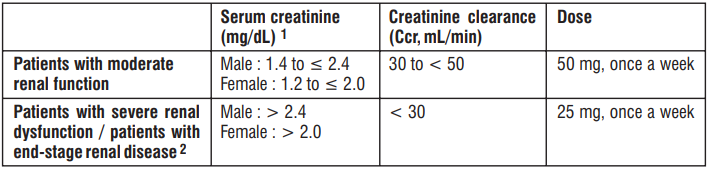
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
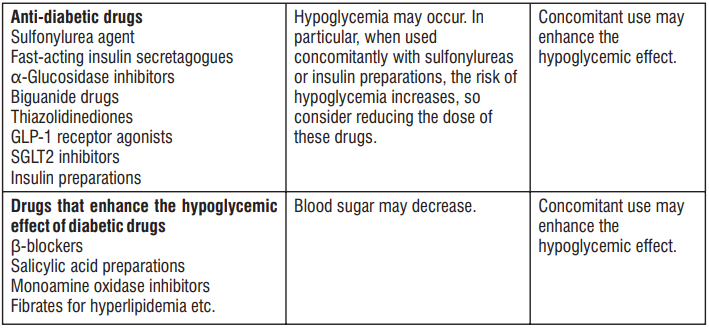

- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.

Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.

In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.

Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025
For More Information About This Product
Trelagliptin Tablets 50 mg
1.0 Generic Name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.
In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.
Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025
For More Information About This Product
Trelagliptin Tablets 25 mg
1.0 Generic name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.
In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.
Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025