Trelagliptin Tablets 100 mg


1.0 Generic Name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
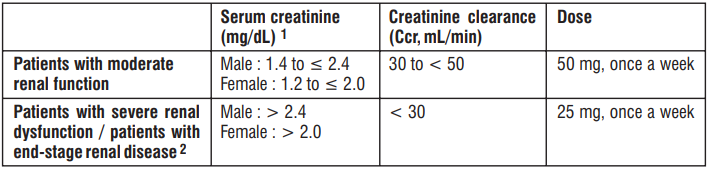
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
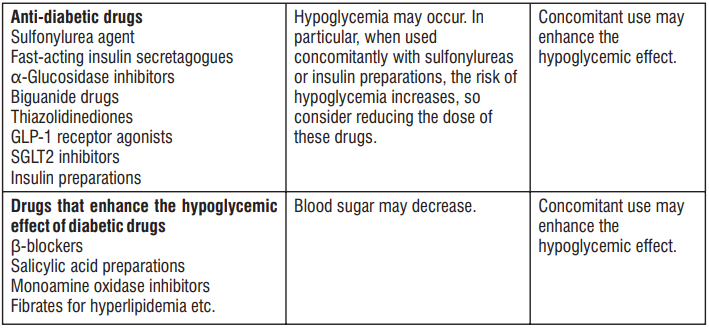

- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.

Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.

In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.

Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
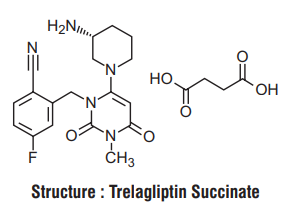
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025
For More Information About This Product
Trelagliptin Tablets 50 mg
1.0 Generic Name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.
In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.
Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025
For More Information About This Product
Trelagliptin Tablets 25 mg
1.0 Generic name
Trelagliptin Tablets 25 mg / 50 mg / 100 mg
2.0 Qualitative and quantitative composition
Trelaglip 25
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 25 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow)and Titanium Dioxide IP
Trelaglip 50
Each film coated tablet contains :
Trelagliptin Succinate equivalent to Trelagliptin 50 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
Trelaglip 100
Each film coated tablet contains :
Trelagliptin Succinate
equivalent to Trelagliptin 100 mg
Excipients q.s.
Colours : Ferric Oxide USP NF (Yellow) and Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 25 mg / 50 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indications
For the treatment of type 2 diabetes mellitus.
4.2 Posology and method of administration
The usual adult dosage is 100 mg of Trelaglip administered orally once a week.
Patients with moderate or severe renal dysfunction
The blood concentration of this drug increases due to delayed excretion; therefore, the dose should be reduced according to the degree of renal function, referring to the table below.
Dosage for patients with moderate or severe renal impairment
1 Conversion value equivalent to CCR (age 60 years, weight 65 kg).
2 For patients with end-stage renal disease, the time relationship between administration of this drug and haemodialysis does not matter.
Elderly
Carefully administer the drug while paying attention to the occurrence of side effects and carefully observing the course. In general, renal function is often impaired.
Method of administration
- Trelaglip is to be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose at the time you notice it and then take it on a predetermined day of the week.
4.3 Contraindications
- Patients with severe ketosis, diabetic coma or precoma, Type 1 diabetes.
- Patients with severe infections, those undergoing surgery or those with severe trauma.
- Patients with a history of hypersensitivity to the ingredients of this drug.
4.4 Special warnings and precautions for use
- This drug may cause hypoglycemia. When using this drug, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be exercised.
- Acute pancreatitis may occur, so instruct patients to consult a doctor immediately if they experience initial symptoms such as persistent severe abdominal pain or vomiting.
- This drug is orally administered once a week and its action continues even after discontinuation of administration. Therefore, pay close attention to the blood glucose level and the occurrence of side effects. In addition, when using other diabetic drugs after discontinuation of this drug, the start time and dose of the drug should be examined based on the blood glucose control status.
- During administration of this drug, blood glucose levels should be checked regularly and the patient's condition should be closely monitored. If the patient shows no satisfactory response after 2 to 3 months of administration of this drug, consideration should be given to changing to a more appropriate treatment.
- Trelaglip and GLP-1 receptor agonists have a blood glucose lowering effect via the GLP-1 receptor. There are no clinical trial results when the two drugs are used together and their efficacy and safety have not been confirmed.
4.5 Drug Interactions
- When Trelagliptin was used in combination with Glimepiride or Metformin, no clear effect was observed on the pharmacokinetics of Trelagliptin and these concomitant drugs.
- When Trelagliptin was used in combination with Caffeine, Tolbutamide, Dextromethorphan or Midazolam, no clear effect was observed on the pharmacokinetics of these concomitant drugs.
4.6 Use in special populations (such as pregnant women, lactating women, paediatric patients, geriatric patients etc.)
Patients with complications / medical history
Patients or conditions at risk of hypoglycemia :
- Pituitary or adrenal insufficiency.
- Malnutrition, starvation, irregular food intake, insufficient food intake or debilitation.
- Intense muscle exercise.
- People who consume excessive amounts of alcohol. Patients with a history of abdominal surgery or intestinal obstruction may cause intestinal obstruction.
Patients with moderate or severe renal dysfunction
Reduce the dose and carefully observe the patient's condition. The blood concentration of this drug increases due to delayed excretion depending on the degree of renal function.
Pregnant women
Pregnant women or women who may be pregnant should only be administered if the therapeutic benefits are judged to outweigh the risks. Placental crossing of drug has been reported in animal studies (rats).
Lactating women
Consider whether to continue or discontinue breast-feeding, taking into account the therapeutic benefits and the benefits of breast-feeding. Animal studies (rats) have shown that the drug is excreted in breast milk.
Pediatric population
No clinical trials have been conducted in children.
Elderly
Pay attention to the occurrence of side effects and administer with caution while closely monitoring the progress. Renal function is generally reduced in many cases.
4.7 Effects on ability to drive and use machines
Hypoglycemic symptoms may occur, so caution should be exercised when administering to patients engaged in activities such as working at heights or driving a car.
4.8 Undesirable effects
Serious side effects
Since the following side effects may appear, observe them thoroughly and if any abnormalities are found, take appropriate measures such as discontinuing administration.
Hypoglycemia (Less than 0.1 - 5%)
Hypoglycemia may occur. When used concomitantly with sulfonylurea agents or insulin preparations, severe hypoglycemic symptoms, including loss of consciousness, have been reported. If hypoglycemic symptoms are observed, appropriate measures should be taken, such as having the patient ingest foods containing carbohydrates. However, glucose should be administered when used concomitantly with an α-glucosidase inhibitor.
Pemphigoid (incidence unknown)
If blisters or erosions appear, consult with a dermatologist and take appropriate measures, such as discontinuing administration. Acute pancreatitis (incidence unknown) If any abnormalities such as persistent severe abdominal pain or vomiting are observed, discontinue administration and take appropriate measures.
Bowel obstruction (incidence unknown)
If any abnormalities such as severe constipation, abdominal distention, persistent abdominal pain, or vomiting are observed, discontinue administration and take appropriate measures.
Other side effects
The following adverse reactions may occur, so observe closely and if any abnormalities found, discontinue administration or take appropriate measures.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
In clinical studies, QT prolongation has been reported when a single dose of 800 mg of Trelagliptin was administered.
Removal of this drug by hemodialysis is not considered useful.
5.0 Pharmacological properties
5.1 Mechanism of action
Trelagliptin inhibits the activity of Dipeptidyl Peptidase-4 (DPP-4), which inactivates glucagon-like peptide-1 (GLP-1), which is secreted into the bloodstream from the intestinal tract in response to oral ingestion of a meal, thereby increasing the blood concentration of GLP-1 and promoting insulin secretion from the pancreas in a glucose concentration-dependent manner.
5.2 Pharmacodynamic properties
Inhibitory effects on DPP-4 Trelagliptin selectively inhibited DPP-4 activity in human plasma (IC50 value : 4.2 nmol/L) (in vitro). In order to compare the DPP-4 inhibitory activity of Trelagliptin and Alogliptin, the IC50 were compared under the same conditions (in vitro) and were found to be 1.3 and 5.3 nmol/L respectively. In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered once weekly (before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite dietary and exercise therapy, the mean DPP-4 activity inhibition rate 7 days after the final administration was 77.4% in the 100 mg Trelagliptin. The plasma Trelagliptin concentration estimated to yield 50% inhibition of plasma DPP-4 activity (IC50) was 1.43 ng/mL (4.02 nmol/L).
Increased concentration of active GLP-1
In a placebo-controlled, double-blind, parallel-group comparative study in which 100 mg of Trelagliptin was orally administered (once a week before breakfast) for 12 weeks to type 2 diabetes patients with insufficient blood sugar control despite diet and exercise therapy, active GLP-1 concentrations in a meal loading test 12 weeks after administration were significantly increased compared to the placebo group.
Improvement of glucose tolerance
Trelagliptin was orally administered once to an overnight fasted obese type 2 diabetes model (Wistar fatty rats) and a non-obese type 2 diabetes model (N-STZ-1.5 rats) and a glucose tolerance test in which glucose was orally administered one hour after administration showed that Trelagliptin improved glucose tolerance.
Phase III clinical trial in India
A trial was conducted on Trelaglip to compare the efficacy and safety of once-weekly Trelaglip (Trelagliptin 100 mg) with twice-daily Vildagliptin 50 mg in patients with type 2 diabetes. Non-inferiority was assessed based on the difference in mean change from baseline in HbA1c at the end of the treatment period. The mean change in HbA1c (± SD) was 0.89 (± 1.46) % in the Trelagliptin group and 0.97 (± 1.42) % in the Vildagliptin group. The incidence of adverse effects was lower in the Trelagliptin group (6.67%) compared to the Vildagliptin group (9.17%), with no cases of hypoglycemia reported in either group. Trelaglip was found to be having better safety and similar efficacy to that of Vildagliptin.
5.3 Pharmacokinetic properties
Single dose study
The pharmacokinetic parameters of Trelagliptin in plasma after a single dose of Trelaglip 100 were as follows. The average plasma concentration after 72 hours of administration was 17.233 ng/mL.
In an internationally conducted study, a single oral dose of 100 mg of Trelagliptin was administered orally 30 minutes before the start of breakfast in healthy adults, the changes in plasma concentrations and pharmacokinetic parameters were as follows and the average plasma concentration after 168 hours of administration was 2.1 ng/mL.
Mean value (standard deviation)
Repeated administration
In healthy adults (9 cases), a single oral dose of 100 mg of Trelagliptin was administered once daily 30 minutes before breakfast and three days later, it was administered once daily for 11 days 30 minutes before breakfast. The average values (standard deviation) of Cmax and AUC0-inf on the first day of administration were 544.3 (122.0) ng/mL and 5572.3 (793.2) ng·h/mL, respectively, while the average th values (standard deviation) of Cmax and AUC0-tau on the 14 day of administration were 602.6 (149.5) ng/mL and 5292.9 (613.8) ng·h/mL respectively.
The steady state was achieved after 12-week treatment with once-weekly Trelagliptin. The plasma concentration of unchanged Trelagliptin was 6.062 ng/mL at 7 days after the last dose. No drug accumulation was observed with repeated dosing of Trelagliptin.
Absorption
When 100 mg of Trelagliptin was orally administered to healthy adults (12 subjects) 30 minutes after the start of breakfast, the Cmax and AUC(0-inf) increased by 16.8% and decreased by 2.5% respectively, compared to when it was administered without breakfast.
Distribution
14 [ C] When Trelagliptin was added to human plasma at a concentration of 0.1 to 10 μg/mL, the protein binding rate was 22.1 to 27.6% (in vitro).
Metabolism
Trelagliptin is metabolized to the active metabolite MI, mainly via N -demethylation by CYP2D6. The amount of the active metabolite MI in human plasma was less than 1% of the amount of unchanged Trelagliptin. Trelagliptin exhibited weak inhibitory effects on CYP3A4/5 (direct inhibitory effect IC50 value : 100 μmol/L or more, metabolic inhibitory effect IC50 values : 12 μmol/L (midazolam 1'-hydroxylation activity) and 28 μmol/L (testosterone 6β-hydroxylation activity)), but did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and did not induce CYP1A2, CYP2B6 or CYP3A4 (in vitro).
Excretion
When a single oral dose of 100 mg of Trelagliptin was administered to 12 healthy adults during breakfast fasting or 30 minutes after the start of breakfast, the cumulative urinary excretion rates of Trelagliptin up to 168 hours were 76.6% and 76.1% respectively. Trelagliptin is a substrate of P-glycoprotein and slightly inhibited the transport of Digoxin through P-glycoprotein (IC50 value : ≥ 500 μmol/L). In addition, Trelagliptin showed an inhibitory effect on the uptake of Metformin, which is a substrate of the organic cation transporter OCT2 (IC50 value : 55.9 μmol/L) (in vitro).
Patients with impaired renal function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with renal impairment and healthy adults, the AUC(0-tlqc) and Cmax were increased by 55.7% and 36.3% in patients with mild renal impairment (Ccr = 50 - 80 mL/min, 6 cases), 105.7% and 12.9% in patients with moderate renal impairment (Ccr = 30 - 50 mL/min, 6 cases), 201.4% and 9.1% in patients with severe renal impairment (Ccr < 30 mL/min, 6 cases), and 268.1% and 13.8% in patients with end-stage renal failure (6 cases) respectively, compared with healthy adults matched for age, sex, race and weight.
In addition, 9.2% of the administered dose of Trelagliptin was removed by 4 hours of hemodialysis.
Patients with impaired hepatic function
When a single dose of 50 mg of Trelagliptin was orally administered to patients with moderate hepatic impairment (Child-Pugh score 7-9, 8 cases) and healthy adults (8 cases), the AUC(0-inf) and Cmax were increased by 5.1% and decreased by 4.3% respectively, compared to healthy adults matched for age, sex, race, smoking history and weight.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Toxicity studies of Trelagliptin included single-dose toxicity, repeated-dose toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity and other toxicity studies (phototoxicity study, skin toxicity studies).
Single dose toxicity
A single-dose toxicity study of Trelaglip was conducted in male and female Mus musculus mice and Wistar rats, with oral doses of 0 (vehicle), 10, 25, 50, 100 or 200 mg/kg body weight. No mortality, biochemical or hematological changes or signs of acute systemic toxicity were observed at the highest dose of 200 mg/kg. An international single-dose toxicity study was performed in male and female Sprague-Dawley (SD) rats with oral doses of 0 (vehicle), 600 or 2000 mg/kg body weight. Salivation was observed immediately after administration and no deaths occurred. The approximate lethal dose of Trelagliptin was determined to be > 2000 mg/kg. In a dose escalation study, escalating single oral doses of Trelagliptin 0 (control), 30, 300 and 2000 mg/kg were administered to male and female beagle dogs. Vomiting, erythema and swelling of the auricle and face, salivation and increases in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase and alkaline phosphatase (ALP) were observed in animals receiving ≥ 300 mg/kg and a decrease in locomotor activity, lateral position and decreased body temperature were observed in animals receiving 2000 mg/kg but no deaths were observed. Thus, the approximate lethal dose was determined to be > 2000 mg/kg.
Repeated dose toxicity
Four-week repeated oral dose toxicity study in mice and rabbits
A four-week repeated-dose toxicity study of Trelaglip was conducted in male and female Swiss albinomice (0, 20.41, 102.9, 204.1 mg/kg) and New Zealand rabbits (0, 5.2, 25.7, 51.46 mg/kg), followed by a recovery phase in the vehicle and high-dose groups to evaluate toxicity reversibility after a two-week recovery period. No adverse effects were observed on growth, body weight or feed consumption in the test animals. While slight elevations in neutrophil and monocyte levels were observed, these remained within the normal laboratory range. Hematological and biochemical parameters showed no signifcant deviations from controls. Any external or internal pathological ndings were not observed after necropsy and gross examination.
Four-week repeated oral dose toxicity study in rats
Male and female SD rats orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 50, 250 or 1000 mg/kg/day for 4 weeks. In addition, a recovery study was conducted in the 0 and 1000 mg/kg/day groups to assess the reversibility of toxicity after a 2-week recovery period. Stained perianogenital fur; increase in neutrophil, lymphocyte and white blood cell counts; increase in inorganic phosphorus, total cholesterol and ALP; decrease in sodium, chloride, albumin and total protein : a trend towards increased urine protein; an increase in liver weight; a decrease in thymus weight; centrilobular hepatocellular hypertrophy and a decrease in lymphocytes in the thymic cortex were observed in the 1000 mg/kg/day group.
All findings were reversible after the 2-week recovery period.
As described above, stained fur associated with worsening of clinical signs and increased white blood cell count suggestive of inflammation were observed in the 1000 mg/kg/day group and therefore the no observed adverse effect level (NOAEL) was determined to be 250 mg/kg/day.
Genotoxicity Bacterial reverse mutation assay, gene mutation assay with mouse lymphoma (L5178Y/TK+/-) and mouse bone marrow micronucleus assay were conducted in a study. The result showed Trelagliptin to have no genotoxicity. Carcinogenecity Carcinogenicity dose-range finding study Male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 100, 300, 1000 or 2000 mg/kg/day for 1 week. Death occurred in 1 of 10 females in the 100 mg/kg/day group, 1 of 10 females in the 300 mg/kg/day group, 2 of 10 males and 1 of 10 females in the 1000 mg/kg/day group and 1 of 10 males and 1 of 10 females in the 2000 mg/kg/day group. In addition, among animals evaluated for toxicokinetics, 1 of 21 females died in the 1000 mg/kg/day group and 1 of 21 males and 7 of 21 females died in the 2000 mg/kg/day group. The deaths observed in the 2000 mg/kg/day group were considered attributable to Trelagliptin because of the high incidence of death. Findings in the 2000 mg/kg/day group were a decrease in locomotor activity, convulsion, abdominal distension, unkept fur, ataxia, decreased food consumption, decrease in lymphocyte and white blood cell counts and increase in ALP, ALT, AST and urea nitrogen.
Separately, an additional group of male and female ICR mice orally received daily 1000 mg/kg/day of Trelagliptin for 1 week.
A decrease in locomotor activity, prone position, bradypnea and hypothermia were observed after the first dose, but not after the second or subsequent doses. In addition, deaths or effects on body weight associated with Trelagliptin were not noted.
Furthermore, male and female ICR mice orally received daily Trelagliptin at 0 (vehicle-0.5% Methylcellulose solution), 150, 300, 600 or 1200 mg/kg/day for 13 weeks. No deaths associated with Trelagliptin occurred, nor were any toxicological findings observed in clinical signs, body weight, food consumption, ophthalmology, haematology, clinical chemistry, organ weights, necropsy or histopathology.
7.0 Description
Trelaglip (Trelagliptin Succinate) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).
Chemical name : R 22-({6-[(3 )-3-Aminopiperidin-1-yl] H -3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1 (2 ) -yl} methyl) -4-fluorobenzonitrile monosuccinate
Molecular formula : C18H20FN5O2.C4H6O4
Molecular weight : 475.47 g/mol
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
A wallet pack of 4 tablets.
8.4 Storage and handling instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients on the following points when administering the drug.
- This drug should be taken once a week and should be taken on the same day of the week.
- If you forget to take this drug, take only the prescribed dose when you notice it and then take it on the predetermined day of the week.
- Since there is a risk of hypoglycemia, patients should be fully informed of the symptoms of hypoglycemia and how to deal with them and caution should be called to them before using this drug.
- Patients should be instructed to seek immediate medical attention if they experience initial symptoms such as persistent severe abdominal pain or vomiting, as acute pancreatitis may occur.
- Since this drug is to be administered orally once a week and the effect continues even after discontinuation, pay close attention to blood glucose levels and the occurrence of side effects. In addition, when using other diabetes drugs after discontinuing administration of this drug, consider the timing and dose of administration based on the status of blood glucose management.
- During the course of administration of the drug, blood glucose should be tested regularly, the progress should be monitored thoroughly and if the effect of the drug is insufficient after 2 to 3 months, a change to a more appropriate treatment should be considered.
- Caution should be exercised when administering to patients who are engaged in high-altitude work, driving a car etc., as it may cause hypoglycemic symptoms.
12.0 Date of issue
06 January 2025
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
- What Trelaglip Tablet is and what it is used for
- What you need to know before you take Trelaglip Tablet
- How to take Trelaglip Tablet
- Possible side effects
- How to store Trelaglip Tablet
- Contents of the pack and other information
1. What Trelaglip Tablet is and what it is used for
Trelaglip contains a medicine called Trelagliptin Succinate. It is used to treat type 2 diabetes in adults. Trelaglip helps control blood sugar levels by increasing the amount of insulin your body makes after meals.
2. What you need to know before you take Trelaglip Tablet
Do not take Trelaglip if you:
- Have severe diabetes complications like diabetic coma or type 1 diabetes.
- Have severe infections, are having surgery, or have severe injuries.
- Are allergic to Trelagliptin or any other ingredients in this medicine.
Warnings and precautions:
- Trelaglip can cause low blood sugar (hypoglycemia). Know the symptoms and how to treat them.
- It can cause inflammation of the pancreas (pancreatitis). Seek medical help if you have severe stomach pain or vomiting.
- Regularly check your blood sugar levels and tell your doctor if there is no improvement after 2-3 months.
Patients with complications or medical history
At risk of low blood sugar (hypoglycemia):
- Problems with your pituitary or adrenal glands.
- Poor nutrition, not eating enough, irregular eating habits, or being very weak.
- Intense physical exercise.
- Drinking too much alcohol.
- If you have had abdominal surgery or have a history of intestinal blockage.
Children and adolescents
Trelaglip is not recommended for children and teenagers under 18 years old.
Pregnancy and breastfeeding
If you are pregnant, breastfeeding, or planning to have a baby, ask your doctor for advice before taking this medicine.
Elderly patients
If you are elderly, your doctor will monitor you closely for side effects, as kidney function may be reduced.
Driving and using machines:
Trelaglip can cause low blood sugar. Do not drive or use machines if you feel symptoms of low blood sugar.
Other drugs with Trelaglip tablet
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes medicines obtained without a prescription, vitamins, and herbal supplements.
Trelaglip may interact with
- Sulfonylureas or insulin: These medicines can increase the risk of low blood sugar. Your doctor may need to adjust the dose.
- Fast-acting insulin secretagogues: These can also increase the risk of low blood sugar.
- Alpha-glucosidase inhibitors: These can enhance the blood sugar-lowering effect.
- Biguanides (e.g., Metformin): No significant interaction observed, but always inform your doctor.
- Thiazolidinediones: These can enhance the blood sugar-lowering effect.
- GLP-1 receptor agonists: No clinical trial results are available for combined use.
- SGLT2 inhibitors: These can enhance the blood sugar-lowering effect.
- Beta-blockers: These can mask the symptoms of low blood sugar.
- Salicylic acid preparations: These can enhance the blood sugar-lowering effect.
- Monoamine oxidase inhibitors: These can enhance the blood sugar-lowering effect.
- Fibrates for hyperlipidemia: These can enhance the blood sugar-lowering effect.
- Corticosteroids, adrenaline, thyroid hormones: These can increase blood sugar levels.
3. How to take Trelaglip Tablet
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
The usual dose is:
- 100 mg once a week, taken by mouth.
For patients with kidney problems:
The dose should be adjusted based on kidney function. Follow your doctor's instructions.
Doses for patients with kidney problems:
- Moderate kidney problems (Creatinine clearance 30 to < 50 mL/min): 50 mg once a week.
- Severe kidney problems (Creatinine clearance < 30 mL/min) and end-stage kidney disease: 25 mg once a week.
If you use more Trelaglip Tablet than you should
Tell your doctor if you accidentally use more than you were told.
If you forget to use Trelaglip Tablet If you forget to take at the right time, use it as soon as you remember, then carry on as before. Do not take a double dose to make up for a forgotten dose.
If you stop using Trelaglip Tablet
Do not stop your treatment even if you feel better unless told to do so by your doctor. If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, Trelaglip can cause side effects, but not everyone gets them.
Serious side effects:
- Low blood sugar (hypoglycemia)
- Inflammation of the pancreas (severe stomach pain or vomiting)
- Bowel blockage (severe constipation, stomach swelling, persistent stomach pain, or vomiting)
Other side effects:
- Rash, itching
- Increased liver enzymes
- Common cold symptoms
Reporting of side effects If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Drug Safety Reporting” located on the top end of the home page. Website link https://www.zuventus.com/drug-safety-reporting .
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Trelaglip Tablet
Store below 30°C. Protect from moisture.
Keep out of reach of children.
6. Contents of the pack and other information
What Trelaglip contains:
- The active ingredient is Trelagliptin Succinate.
- Other ingredients include Ferric Oxide (Yellow), Titanium Dioxide, and other excipients.
What Trelaglip looks like and contents of the pack:
- Trelaglip comes in tablet form in strengths of 25 mg, 50 mg, and 100 mg.
- The tablets are packed in a wallet pack of 4 tablets.
This leaflet was last revised on: 06 February 2025
For More Information About This Product
Vil GM 1000 Tablets
1.0 Generic name
Vildagliptin & Metformin Hydrochloride Tablets IP
2.0 Qualitative and quantitative composition
Vil GM 500
Each film coated tablet contains :
Vildagliptin IP 50 mg
Metformin Hydrochloride IP 500 mg
Excipients q.s.
Colours : Yellow Oxide of Iron & Titanium Dioxide IP
Vil GM 1000
Each film coated tablet contains :
Vildagliptin IP 50 mg
Metformin Hydrochloride IP 1000 mg
Excipients
Colours : Yellow Oxide of Iron & Titanium Dioxide IP
3.0 Dosage form and strength
Tablet, 50 mg / 500 mg and 50 mg / 1000 mg
4.0 Clinical particulars
4.1 Therapeutic indication
For the treatment of type-II diabetes mellitus when single drug therapy along with diet, exercise do not result in adequate glycemic control.
4.2 Posology and method of administration
Adults with normal renal function (GFR ≥ 90 ml/min) The dose of antihyperglycaemic therapy with Vil GM should be individualised on the basis of the patient's current regimen, effectiveness and tolerability while not exceeding the maximum recommended daily dose of 100 mg Vildagliptin.
Vil GM may be initiated at either the 50 mg / 500 mg or 50 mg / 1000 mg tablet strength twice daily, one tablet in the morning and the other in the evening. For patients inadequately controlled at their maximal tolerated dose of Metformin monotherapy :
The starting dose of Vil GM should provide Vildagliptin as 50 mg twice daily (100 mg total daily dose) plus the dose of Metformin already being taken. For patients switching from co-administration of Vildagliptin and Metformin as separate tablets :
Vil GM should be initiated at the dose of Vildagliptin and Metformin already being taken. For patients inadequately controlled on dual combination with Metformin and a Sulphonylurea :
The doses of Vil GM should provide Vildagliptin as 50 mg twice daily (100 mg total daily dose) and a dose of Metformin similar to the dose already being taken. When Vil GM is used in combination with a Sulphonylurea, a lower dose of the Sulphonylurea may be considered to reduce the risk of hypoglycaemia. For patients inadequately controlled on dual combination therapy with insulin and the maximal tolerated dose of Metformin :
The dose of Vil GM should provide Vildagliptin dosed as 50 mg twice daily (100 mg total daily dose) and a dose of Metformin similar to the dose already being taken. The safety and efficacy of Vildagliptin and Metformin as triple oral therapy in combination with a Thiazolidinedione have not been established.
Special populations
Elderly (≥ 65 years)
As Metformin is excreted via the kidney, and elderly patients have a tendency to decreased renal function, elderly patients taking Vil GM should have their renal function monitored regularly.
Renal impairment
A GFR should be assessed before initiation of treatment with Metformin-containing products and at least annually thereafter. In patients at increased risk of further progression of renal impairment and in the elderly, renal function should be assessed more frequently, e.g. every 3 - 6 months. The maximum daily dose of Metformin should preferably be divided into 2 - 3 daily doses. Factors that may increase the risk of lactic acidosis should be reviewed before considering initiation of Metformin in patients with GFR < 60 ml/min.
If no adequate strength of Vil GM is available, individual monocomponents should be used instead of the fixed dose combination.
Hepatic impairment
Vil GM should not be used in patients with hepatic impairment, including those with pre-treatment alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 3 times the upper limit of normal (ULN).
Paediatric population
Vil GM is not recommended for use in children and adolescents (< 18 years). The safety and efficacy of Vil GM in children and adolescents (< 18 years) have not been established. No data are available.
Method of administration
Oral use
Taking Vil GM with or just after food may reduce gastrointestinal symptoms associated with Metformin
4.3 Contraindications
- Hypersensitivity to the active substances or to any of the excipients.
- Any type of acute metabolic acidosis. (such as lactic acidosis, diabetic ketoacidosis.)
- Diabetic pre-coma
- Severe renal failure (GFR < 30 ml/min)
- Acute conditions with the potential to alter renal function, such as
- Dehydration
- Severe infection
- Shock
- Intravascular administration of iodinated contrast agents
- Acute or chronic disease which may cause tissue hypoxia, such as
- Cardiac or respiratory failure
- Recent myocardial infarction
- Shock
- Hepatic impairment
- Acute alcohol intoxication, alcoholism
- Breast-feeding
4.4 Special warnings and precautions for use
General
Vil GM is not a substitute for insulin in insulin-requiring patients and should not be used in patients with type 1 diabetes
Lactic acidosis
Lactic acidosis, a very rare but serious metabolic complication, most often occurs at acute worsening of renal function, or cardiorespiratory illness or sepsis. Metformin accumulation occurs at acute worsening of renal function and increases the risk of lactic acidosis.
In case of dehydration (severe diarrhoea or vomiting, fever or reduced fluid intake), Metformin should be temporarily discontinued and contact with a health care professional is recommended.
Medicinal products that can acutely impair renal function (such as antihypertensives, diuretics and NSAIDs) should be initiated with caution in Metformin-treated patients. Other risk factors for lactic acidosis are excessive Alcohol intake, hepatic insufficiency, inadequately controlled diabetes, ketosis, prolonged fasting and any conditions associated with hypoxia, as well as concomitant use of medicinal products that may cause lactic acidosis. Patients and/or care-givers should be informed of the risk of lactic acidosis. Lactic acidosis is characterised by acidotic dyspnoea, abdominal pain, muscle cramps, asthenia and hypothermia followed by coma. In case of suspected symptoms, the patient should stop taking Metformin and seek immediate medical attention. Diagnostic laboratory findings are decreased blood pH (< 7.35), increased plasma lactate levels (> 5 mmol/l) and an increased anion gap and lactate/pyruvate ratio.
Administration of iodinated contrast agents
Intravascular administration of iodinated contrast agents may lead to contrast-induced nephropathy, resulting in Metformin accumulation and increased risk of lactic acidosis. Metformin should be discontinued prior to or at the time of the imaging procedure and not restarted until at least 48 hours after, provided that renal function has been reevaluated and found to be stable
Renal function
GFR should be assessed before treatment initiation and regularly thereafter. Metformin is contraindicated in patients with GFR < 30 ml/min and should be temporarily discontinued in the presence of conditions that alter renal function.
Hepatic impairment
Patients with hepatic impairment, including those with pre-treatment ALT or AST > 3x ULN, should not be treated with Vil GM
Liver enzyme monitoring
Rare cases of hepatic dysfunction (including hepatitis) have been reported with Vildagliptin. In these cases, the patients were generally asymptomatic without clinical sequelae and liver function tests (LFTs) returned to normal after discontinuation of treatment. LFTs should be performed prior to the initiation of treatment with Vil GM in order to know the patient's baseline value. Liver function should be monitored during treatment with Vil GM at three-month intervals during the first year and periodically thereafter. Patients who develop increased transaminase levels should be monitored with a second liver function evaluation to confirm the finding and be followed thereafter with frequent LFTs until the abnormality(ies) return(s) to normal. Should an increase in AST or in ALT of 3x ULN or greater persist, withdrawal of Vil GM therapy is recommended. Patients who develop jaundice or other signs suggestive of liver dysfunction should discontinue Vil GM. Following withdrawal of treatment with Vil GM and LFT normalisation, treatment with Vil GM should not be re-initiated.
Skin disorders
Skin lesions, including blistering and ulceration have been reported with Vildagliptin in extremities of monkeys in nonclinical toxicology studies. Although skin lesions were not observed at an increased incidence in clinical trials, there was limited experience in patients with diabetic skin complications. Furthermore, there have been post-marketing reports of bullous and exfoliative skin lesions. Therefore, in keeping with routine care of the diabetic patient, monitoring for skin disorders, such as blistering or ulceration, is recommended.
Acute pancreatitis
Use of Vildagliptin has been associated with a risk of developing acute pancreatitis. Patients should be informed of the characteristic symptom of acute pancreatitis. If pancreatitis is suspected, Vildagliptin should be discontinued; if acute pancreatitis is confirmed, Vildagliptin should not be restarted. Caution should be exercised in patients with a history of acute pancreatitis.
Hypoglycaemia
Sulphonylureas are known to cause hypoglycaemia. Patients receiving Vildagliptin in combination with a Sulphonylurea may be at risk for hypoglycaemia. Therefore, a lower dose of Sulphonylurea may be considered to reduce the risk of hypoglycaemia.
Surgery
Metformin must be discontinued at the time of surgery under general, spinal or epidural anaesthesia. Therapy may be restarted no earlier than 48 hours following surgery or resumption of oral nutrition and provided that renal function has been re-evaluated and found to be stable.
4.5 Drugs interactions
There have been no formal interaction studies for Vil GM. The following statements reflect the information available on the individual active substances.
Vildagliptin
- Vildagliptin has a low potential for interactions with co-administered medicinal products. Since Vildagliptin is not a cytochrome P (CYP) 450 enzyme substrate and does not inhibit or induce CYP 450 enzymes, it is not likely to interact with active substances that are substrates, inhibitors or inducers of these enzymes.
- Results from clinical trials conducted with the oral antidiabetics Pioglitazone, Metformin and Glyburide in combination with Vildagliptin have shown no clinically relevant pharmacokinetic interactions in the target population.
- Drug-drug interaction studies with Digoxin (P-glycoprotein substrate) and Warfarin (CYP2C9 substrate) in healthy subjects have shown no clinically relevant pharmacokinetic interactions after co-administration with Vildagliptin.
- Drug-drug interaction studies in healthy subjects were conducted with Amlodipine, Ramipril, Valsartan and Simvastatin. In these studies, no clinically relevant pharmacokinetic interactions were observed after coadministration with Vildagliptin. However, this has not been established in the target population.
- Combination with ACE inhibitors.
- There may be an increased risk of angioedema in patients concomitantly taking ACE inhibitors.
- As with other oral antidiabetic medicinal products the hypoglycaemic effect of Vildagliptin may be reduced by certain active substances, including Thiazides, Corticosteroids, Thyroid products and Sympathomimetics.
Metformin
Combinations not recommended
- Alcohol Alcohol intoxication is associated with an increased risk of lactic acidosis, particularly in cases of fasting, malnutrition or hepatic impairment.
- Iodinated contrast agents Metformin must be discontinued prior to or at the time of the imaging procedure and not restarted until at least 48 hours after, provided that renal function has been re-evaluated and found to be stable.
- Cationic active substances Cationic active substances that are eliminated by renal tubular secretion (e.g. Cimetidine) may interact with Metformin by competing for common renal tubular transport systems and hence delay the elimination of Metformin, which may increase the risk of lactic acidosis. A study in healthy volunteers showed that Cimetidine, administered as 400 mg twice daily, increased Metformin systemic exposure (AUC) by 50%. Therefore, close monitoring of glycaemic control, dose adjustment within the recommended posology and changes in diabetic treatment should be considered when cationic medicinal products that are eliminated by renal tubular secretion are co-administered.
Combinations requiring precautions for use
- Some medicinal products can adversely affect renal function which may increase the risk of lactic acidosis, e.g. NSAIDs, including selective Cyclo-oxygenase (COX) II inhibitors, ACE inhibitors, angiotensin II receptor antagonists and diuretics, especially loop diuretics. When starting or using such products in combination with Metformin, close monitoring of renal function is necessary.
- Glucocorticoids, beta-2-agonists and diuretics have intrinsic hyperglycaemic activity. The patient should be informed and more frequent blood glucose monitoring performed, especially at the beginning of treatment. If necessary, the dosage of Vil GM may need to be adjusted during concomitant therapy and on its discontinuation.
- Angiotensin converting enzyme (ACE) inhibitors may decrease the blood glucose levels. If necessary, the dosage of the antihyperglycaemic medicinal product should be adjusted during therapy with the other medicinal product and on its discontinuation.
4.6 Use in special populations
Pregnancy
There are no adequate data from the use of Vil GM in pregnant women. For Vildagliptin studies in animals have shown reproductive toxicity at high doses. For Metformin, studies in animals have not shown reproductive toxicity. Studies in animals performed with Vildagliptin and Metformin have not shown evidence of teratogenicity, but foetotoxic effects at maternotoxic doses. The potential risk for humans is unknown. Vil GM should not be used during pregnancy
Nursing mothers Studies in animals have shown excretion of both Metformin and Vildagliptin in milk. It is unknown whether Vildagliptin is excreted in human milk, but Metformin is excreted in human milk in low amounts. Due to both the potential risk of neonate hypoglycaemia related to Metformin and the lack of human data with Vildagliptin, Vil GM should not be used during breast-feeding
Fertility No studies on the effect on human fertility have been conducted for Vil GM.
4.7 Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. Patients who may experience dizziness as an adverse reaction should avoid driving vehicles or using machines.
4.8 Undesirable effects
Arthralgia is reported with Vildagliptin in recent reports. Adverse reactions reported in patients who received Vildagliptin and Metformin in double-blind studies are listed below by system organ class and absolute frequency. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
- Metabolism and nutrition disorders
Common - Hypoglycaemia
- Nervous system disorders
Common - Tremor, Headache, Dizziness
Uncommon - Fatigue
- Gastrointestinal disorders
Common - Nausea
In controlled clinical trials with the combination of Vildagliptin 100 mg daily plus Metformin, no withdrawal due to adverse reactions was reported in either the Vildagliptin 100 mg daily plus Metformin or the placebo plus Metformin treatment groups. Clinical trials of up to more than 2 years' duration did not show any additional safety signals or unforeseen risks when Vildagliptin was added on to Metformin.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com Website : https://www.zuventus.com/drug-safety-reporting
4.9 Overdose
Symptoms
Vildagliptin
Information regarding overdose with Vildagliptin is limited. Information on the likely symptoms of overdose was taken from a rising dose tolerability study in healthy subjects given Vildagliptin for 10 days. At 400 mg, there were three cases of muscle pain, and individual cases of mild and transient paraesthesia, fever, oedema and a transient increase in lipase levels. At 600 mg, one subject experienced oedema of the feet and hands, and increases in creatine phosphokinase (CPK), aspartate aminotransferase (AST), Creactive protein (CRP) and myoglobin levels. Three other subjects experienced oedema of the feet, with paraesthesia in two cases. All symptoms and laboratory abnormalities resolved without treatment after discontinuation of the study medicinal product.
Metformin
A large overdose of Metformin (or co-existing risk of lactic acidosis) may lead to lactic acidosis, which is a medical emergency and must be treated in hospital.
Management
The most effective method of removing Metformin is haemodialysis. However, Vildagliptin cannot be removed by haemodialysis, although the major hydrolysis metabolite (LAY 151) can. Supportive management is recommended.
5.0 Pharmacological properties
5.1 Mechanism of action
Vil GM combines two antihyperglycaemic agents with complimentary mechanisms of action to improve glycaemic control in patients with type 2 diabetes : Vildagliptin, a member of the islet enhancer class, and Metformin Hydrochloride, a member of the biguanide class. The administration of Vildagliptin results in a rapid and complete inhibition of DPP-4 activity, resulting in increased fasting and postprandial endogenous levels of the incretin hormones GLP-1 (Glucagon-Like Peptide 1) and GIP (Glucose-dependent Insulinotropic Polypeptide). Metformin acts primarily by decreasing endogenous hepatic glucose production.
5.2 Pharmacodynamic properties
Vildagliptin
By increasing the endogenous levels of these incretin hormones, Vildagliptin enhances the sensitivity of beta cells to glucose, resulting in improved glucose-dependent insulin secretion. Treatment with Vildagliptin 50 - 100 mg daily in patients with type 2 diabetes significantly improved markers of beta cell function including HOMA-β (Homeostasis Model Assessment-β), proinsulin to insulin ratio and measures of beta cell responsiveness from the frequentlysampled meal tolerance test. In non-diabetic (normal glycaemic) individuals, Vildagliptin does not stimulate insulin secretion or reduce glucose levels.
By increasing endogenous GLP-1 levels, Vildagliptin also enhances the sensitivity of alpha cells to glucose, resulting in more glucose-appropriate glucagon secretion. The enhanced increase in the insulin/glucagon ratio during hyperglycaemia due to increased incretin hormone levels results in a decrease in fasting and postprandial hepatic glucose production, leading to reduced glycaemia. The known effect of increased GLP-1 levels delaying gastric emptying is not observed with Vildagliptin treatment.
Metformin
Metformin is a biguanide with antihyperglycaemic effects, lowering both basal and postprandial plasma glucose. It does not stimulate insulin secretion and therefore does not produce hypoglycaemia or increased weight gain. Metformin may exert its glucose-lowering effect via three mechanisms :
- By reduction of hepatic glucose production through inhibition of gluconeogenesis and glycogenolysis.
- In muscle, by modestly increasing insulin sensitivity, improving peripheral glucose uptake and utilisation.
- By delaying intestinal glucose absorption.
Metformin stimulates intracellular glycogen synthesis by acting on glycogen synthase and increases the transport capacity of specific types of membrane glucose transporters (GLUT-1 and GLUT-4). In humans, independently of its action on Glycaemia, Metformin has favourable effects on lipid metabolism.
5.3 Pharmacokinetic properties
Vil GM
Bioequivalence has been demonstrated between Vil GM at three dose strengths (50 mg / 500 mg, 50 mg / 850 mg and 50 mg / 1000 mg) versus free combination of Vildagliptin and Metformin Hydrochloride tablets at the corresponding doses. Food does not affect the extent and rate of absorption of Vildagliptin from Vil GM. The rate and extent of absorption of Metformin from Vil GM 50 mg / 1000 mg were decreased when given with food as reflected by the decrease in Cmax by 26%, AUC by 7% and delayed Tmax (2.0 to 4.0 h). The following statements reflect the pharmacokinetic properties of the individual active substances of Vil GM.
Vildagliptin
Absorption
Following oral administration in the fasting state, Vildagliptin is rapidly absorbed, with peak plasma concentrations observed at 1.7 hours. Food slightly delays the time to peak plasma concentration to 2.5 hours, but does not alter the overall exposure (AUC). Administration of Vildagliptin with food resulted in a decreased Cmax (19%). However, the magnitude of change is not clinically significant, so that Vildagliptin can be given with or without food. The absolute bioavailability is 85%.
Distribution
The plasma protein binding of Vildagliptin is low (9.3%) and Vildagliptin distributes equally between plasma and red blood cells. The mean volume of distribution of Vildagliptin at steady-state after intravenous administration (Vss) is 71 litres, suggesting extravascular distribution.
Biotransformation
Metabolism is the major elimination pathway for Vildagliptin in humans, accounting for 69% of the dose. The major metabolite (LAY 151) is pharmacologically inactive and is the hydrolysis product of the cyano moiety, accounting for 57% of the dose, followed by the glucuronide (BQS867) and the amide hydrolysis products (4% of dose). In vitro data in human kidney microsomes suggest that the kidney may be one of the major organs contributing to the hydrolysis of Vildagliptin to its major inactive metabolite, LAY151. DPP-4 contributes partially to the hydrolysis of Vildagliptin based on an in vivo study using DPP-4 deficient rats. Vildagliptin is not metabolised by CYP 450 enzymes to any quantifiable extent. Accordingly, the metabolic clearance of Vildagliptin is not anticipated to be affected by co-medications that are CYP 450 inhibitors and/or inducers. In vitro studies demonstrated that Vildagliptin does not inhibit/induce CYP 450 enzymes. Therefore, Vildagliptin is not likely to affect metabolic clearance of co-medications metabolised by CYP 1A2, CYP 2C8, CYP 2C9, CYP 2C19, CYP 2D6, CYP 2E1 or CYP 3A4/5.
Elimination
Following oral administration of [14C] Vildagliptin, approximately 85% of the dose was excreted into the urine and 15% of the dose is recovered in the faeces. Renal excretion of the unchanged Vildagliptin accounted for 23% of the dose after oral administration. After intravenous administration to healthy subjects, the total plasma and renal clearances of Vildagliptin are 41 and 13 l/h, respectively. The mean elimination half-life after intravenous administration is approximately 2 hours. The elimination half-life after oral administration is approximately 3 hours.
Metformin
Absorption
After an oral dose of Metformin, the maximum plasma concentration (Cmax) is achieved after about 2.5 h. Absolute bioavailability of a 500 mg Metformin tablet is approximately 50 - 60% in healthy subjects. After an oral dose, the nonabsorbed fraction recovered in faeces was 20 - 30%. After oral administration, Metformin absorption is saturable and incomplete. It is assumed that the pharmacokinetics of Metformin absorption are non-linear. At the usual Metformin doses and dosing schedules, steady state plasma concentrations are reached within 24 - 48 h and are generally less than 1 µg/ml. In controlled clinical trials, maximum Metformin plasma levels (Cmax) did not exceed 4 µg/ml, even at maximum doses. Food slightly delays and decreases the extent of the absorption of Metformin. Following administration of a dose of 850 mg, the plasma peak concentration was 40% lower, AUC was decreased by 25% and time to peak plasma concentration was prolonged by 35 minutes. The clinical relevance of this decrease is unknown.
Distribution
Plasma protein binding is negligible. Metformin partitions into erythrocytes. The mean volume of distribution (Vd) ranged between 63 - 276 litres.
Biotransformation
Metformin is excreted unchanged in the urine. No metabolites have been identified in humans
Elimination
Metformin is eliminated by renal excretion. Renal clearance of Metformin is > 400 ml/min, indicating that Metformin is eliminated by glomerular filtration and tubular secretion. Following an oral dose, the apparent terminal elimination half-life is approximately 6.5 h. When renal function is impaired, renal clearance is decreased in proportion to that of creatinine and thus the elimination half-life is prolonged, leading to increased levels of Metformin in plasma.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Animal studies of up to 13-week duration have been conducted with the combined substances in Vil GM. No new toxicities associated with the combination were identified. The following data are findings from studies per formed with Vildagliptin or Metformin individually.
Vildagliptin
Intra-cardiac impulse conduction delays were observed in dogs with a no-effect dose of 15 mg/kg (7-fold human exposure based on Cmax). Accumulation of foamy alveolar macrophages in the lung was observed in rats and mice. The no-effect dose in rats was 25 mg/kg (5-fold human exposure based on AUC) and in mice 750 mg/kg (142-fold human exposure). Gastrointestinal symptoms, particularly soft faeces, mucoid faeces, diarrhoea and, at higher doses, faecal blood were observed in dogs. A no-effect level was not established. Vildagliptin was not mutagenic in conventional in vitro and in vivo tests for genotoxicity. A fertility and early embryonic development study in rats revealed no evidence of impaired fertility, reproductive performance or early embryonic development due to Vildagliptin. Embryo-foetal toxicity was evaluated in rats and rabbits. An increased incidence of wavy ribs was observed in rats in association with reduced maternal body weight parameters, with a no-effect dose of 75 mg/kg (10-fold human exposure). In rabbits, decreased foetal weight and skeletal variations indicative of developmental delays were noted only in the presence of severe maternal toxicity, with a no-effect dose of 50 mg/kg (9-fold human exposure). A pre- and postnatal development study was performed in rats. Findings were only observed in association with maternal toxicity at ≥ 150 mg/kg and included a transient decrease in body weight and reduced motor activity in the F1 generation. A two-year carcinogenicity study was conducted in rats at oral doses up to 900 mg/kg (approximately 200 times human exposure at the maximum recommended dose). No increases in tumour incidence attributable to Vildagliptin were observed. Another two-year carcinogenicity study was conducted in mice at oral doses up to 1,000 mg/kg. An increased incidence of mammary adenocarcinomas and haemangiosarcomas was observed with a no-effect dose of 500 mg/kg (59-fold human exposure) and 100 mg/kg (16-fold human exposure), respectively. The increased incidence of these tumours in mice is considered not to represent a significant risk to humans based on the lack of genotoxicity of Vildagliptin and its principal metabolite, the occurrence of tumours only in one species and the high systemic exposure ratios at which tumours were observed. In a 13-week toxicology study in cynomolgus monkeys, skin lesions have been recorded at doses ≥ 5 mg/kg/day. These were consistently located on the extremities (hands, feet, ears and tail). At 5 mg/kg/day (approximately equivalent to human AUC exposure at the 100 mg dose), only blisters were observed. They were reversible despite continued treatment and were not associated with histopathological abnormalities. Flaking skin, peeling skin, scabs and tail sores with correlating histopathological changes were noted at doses ≥ 20 mg/kg/day (approximately 3 times human AUC exposure at the 100 mg dose). Necrotic lesions of the tail were observed at ≥ 80 mg/kg/day. Skin lesions were not reversible in the monkeys treated at 160 mg/kg/day during a 4-week recovery period.
Metformin
Non-clinical data on Metformin reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and toxicity to reproduction
7.0 Description
Vil GM tablet combines two antihyperglycaemic agents with complimentary mechanisms of action to improve glycaemic control in patients with type 2 diabetes : Vildagliptin, a member of the islet enhancer class, and Metformin Hydrochloride, a member of the biguanide class.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Vil GM 500 : Alu-Alu blister strip of 10 tablets.
Vil GM 1000 : Alu-Alu blister strip of 10 tablets.
8.4 Storage and handing instructions
Store protected from moisture at a temperature not exceeding 30°C.
Keep out of reach of children.
9.0 Patient counselling information
Do not take Vil GM
- If you are allergic to Vildagliptin, Metformin or any of the other ingredients of this medicine. If you think you may be allergic to any of these, talk to your doctor before taking Vil GM.
- If you have uncontrolled diabetes, with, for example, severe hyperglycaemia (high blood glucose), nausea, vomiting, diarrhoea, rapid weight loss, lactic acidosis or ketoacidosis. Ketoacidosis is a condition in which substances called Ketone bodies accumulate in the blood and which can lead to diabetic pre-coma. Symptoms include stomach pain, fast and deep breathing, sleepiness or your breath developing an unusual fruity smell.
- If you have recently had a heart attack or if you have heart failure or serious problems with your blood circulation or difficulties in breathing which could be a sign of heart problems.
- If you have severely reduced kidney function.
- If you have a severe infection or are seriously dehydrated (have lost a lot of water from your body).
- If you are going to have a contrast x-ray (a specific type of x-ray involving an injectable dye).
- If you have liver problems.
- If you drink alcohol excessively (whether every day or only from time to time).
- If you are breast-feeding.
Warnings and precautions
Risk of lactic acidosis
Vil GM may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration, liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease). If any of the above apply to you, talk to your doctor for further instructions.
Stop taking Vil GM for a short time
If you have a condition that may be associated with dehydration (significant loss of body fluids) such as severe vomiting, diarrhoea, fever, exposure to heat or if you drink less fluid than normal. Talk to your doctor for further instructions. Stop taking Vil GM and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma. Symptoms of lactic acidosis include :
- Vomiting
- Stomach ache (abdominal pain)
- Muscle cramps
- A general feeling of not being well with severe tiredness
- Difficulty in breathing
- Reduced body temperature and heartbeat
Lactic acidosis is a medical emergency and must be treated in a hospital. Vil GM is not a substitute for insulin. Therefore, you should not receive Vil GM for the treatment of type 1 diabetes. Talk to your doctor, pharmacist or nurse before taking Vil GM if you have or have had a disease of the pancreas. Talk to your doctor, pharmacist or nurse before taking Vil GM if you are taking an anti-diabetic medicine known as a sulphonylurea. Your doctor may want to reduce your dose of the sulphonylurea when you take it together with Vil GM in order to avoid low blood glucose (hypoglycaemia). If you have previously taken vildagliptin but had to stop taking it because of liver disease, you should not take this medicine.
Diabetic skin lesions are a common complication of diabetes. You are advised to follow the recommendations for skin and foot care that you are given by your doctor or nurse. You are also advised to pay particular attention to new onset of blisters or ulcers while taking Vil GM. Should these occur, you should promptly consult your doctor. If you need to have major surgery you must stop taking Vil GM during and for some time after the procedure. Your doctor will decide when you must stop and when to restart your treatment with Vil GM. A test to determine your liver function will be performed before the start of Vil GM treatment, at three-month intervals for the first year and periodically thereafter. This is so that signs of increased liver enzymes can be detected as early as possible
During treatment with Vil GM, your doctor will check your kidney function at least once a year or more frequently if you are elderly and/or have worsening renal function. Your doctor will test your blood and urine for sugar regularly.
Children and adolescents
The use of Vil GM in children and adolescents up to 18 years of age is not recommended.
Other medicines and Vil GM
If you need to have an injection of a contrast medium that contains iodine into your bloodstream, for example in the context of an X-ray or scan, you must stop taking Vil GM before or at the time of the injection. Your doctor will decide when you must stop and when to restart your treatment with Vil GM. Tell your doctor if you are taking, have recently taken or might take any other medicines. You may need more frequent blood glucose and kidney function tests, or your doctor may need to adjust the dosage of Vil GM. It is especially important to mention the following :
- Glucocorticoids generally used to treat inflammation.
- Beta-2 agonists generally used to treat respiratory disorders.
- Other medicines used to treat diabetes.
- Medicines which increase urine production (diuretics).
- Medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as Ibuprofen and Celecoxib).
- Certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists).
- Certain medicines affecting the thyroid.
- Certain medicines affecting the nervous system.
- Certain medicines used to treat angina (e.g. Ranolazine).
- Certain medicines used to treat HIV infection (e.g. Dolutegravir).
- Certain medicines used to treat a specific type of thyroid cancer (medullary thyroid cancer) (e.g. Vandetanib).
- Certain medicines used to treat heartburn and peptic ulcers (e.g Cimetidine).
Vil GM with alcohol
Avoid excessive alcohol intake while taking Vil GM since this may increase the risk of lactic acidosis.
Pregnancy and breast-feeding
If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor for advice before taking this medicine. Your doctor will discuss with you the potential risk of taking Vil GM during pregnancy. Do not use Vil GM if you are pregnant or breast-feeding. Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
If you feel dizzy while taking Vil GM, do not drive or use any tools or machines.
When and how to take Vil GM
Swallow the tablets whole with a glass of water. Take one tablet in the morning and the other in the evening with or just after food. Taking the tablet just after food will lower the risk of an upset stomach. Continue to follow any advice about diet that your doctor has given you. In particular, if you are following a diabetic weight control diet, continue with this while you are taking Vil GM.
If you take more Vil GM than you should
If you take too many Vil GM tablets, or if someone else takes your tablets, talk to a doctor or pharmacist immediately. Medical attention may be necessary. If you have to go to a doctor or hospital, take the pack and this leaflet with you.
If you forget to take Vil GM
If you forget to take a tablet, take it with your next meal unless you are due to take one then anyway. Do not take a double dose (two tablets at once) to make up for a forgotten tablet.
If you stop taking Vil GM
Continue to take this medicine as long as your doctor prescribes it so that it can continue to control your blood sugar. Do not stop taking Vil GM unless your doctor tells you to. If you have any questions about how long to take this medicine, talk to your doctor.
12.0 Date of revision
10 November 2022
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What VIL GM is and what it is used for
- What you need to know before you take VIL GM
- How to take VIL GM
- Possible side effects
- How to store VIL GM
- Contents of the pack and other information
1. What Vil Gm is and What It is Used for
The active substances of VIL GM, vildagliptin and metformin, belong to a group of medicines called “oral antidiabetics”. VIL GM is used to treat adult patients with type 2 diabetes. This type of diabetes is also known as noninsulin-dependent diabetes mellitus. Type 2 diabetes develops if the body does not make enough insulin or if the insulin that the body makes does not work as well as it should. It can also develop if the body produces too much glucagon. Both insulin and glucagon are made in the pancreas. Insulin helps to lower the level of sugar in the blood, especially after meals. Glucagon triggers the liver to make sugar, causing the blood sugar level to rise.
How VIL GM works
Both active substances, vildagliptin and metformin, help to control the level of sugar in the blood. The substance vildagliptin works by making the pancreas produce more insulin and less glucagon. The substance metformin works by helping the body to make better use of insulin. This medicine has been shown to reduce blood sugar, which may help to prevent complications from your diabetes.
Even though you are now starting a medicine for your diabetes, it is important that you continue to follow the diet and/or exercise which has been recommended for you.
2. What You Need to Know Before You Take Vil Gm
Do not use VIL GM
if you are allergic to vildagliptin, metformin or any of the other ingredients of this medicine.
If you think you may be allergic to any of these, talk to your doctor before taking VIL GM.
if you have uncontrolled diabetes, with, for example, severe hyperglycaemia (high blood glucose), nausea, vomiting, diarrhoea, rapid weight loss, lactic acidosis or ketoacidosis. Ketoacidosis is a condition in which substances called ketone bodies accumulate in the blood and which can lead to diabetic pre-coma. Symptoms include stomach pain, fast and deep breathing, sleepiness or your breath developing an unusual fruity smell.
if you have recently had a heart attack or if you have heart failure or serious problems with your blood circulation or difficulties in breathing which could be a sign of heart problems.
if you have severely reduced kidney function. if you have a severe infection or are seriously dehydrated (have lost a lot of water from your body).
if you are going to have a contrast x-ray (a specific type of x-ray involving an injectable dye). Please also see information about this in section “Warnings and precautions”.
if you have liver problems. if you drink alcohol excessively (whether every day or only from time to time).
if you are breast-feeding (see also “Pregnancy and breast-feeding”).
Warnings and precautions
Risk of lactic acidosis
VIL GM may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration (see further information below), liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease). If any of the above apply to you, talk to your doctor for further instructions.
Stop taking VIL GM for a short time if you have a condition that may be associated with dehydration (significant loss of body fluids) such as severe vomiting, diarrhoea, fever, exposure to heat or if you drink less fluid than normal. Talk to your doctor for further instructions.
Stop taking VIL GM and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma. Symptoms of lactic acidosis include:
- vomiting
- stomach ache (abdominal pain)
- muscle cramps
- a general feeling of not being well with severe tiredness
- difficulty in breathing
- reduced body temperature and heartbeat
Lactic acidosis is a medical emergency and must be treated in a hospital.
VIL GM is not a substitute for insulin. Therefore, you should not receive VIL GM for the treatment of type 1 diabetes. Talk to your doctor, pharmacist or nurse before taking VIL GM if you have or have had a disease of the pancreas. Talk to your doctor, pharmacist or nurse before taking VIL GM if you are taking an anti-diabetic medicine known as a sulphonylurea. Your doctor may want to reduce your dose of the sulphonylurea when you take it together with VIL GM in order to avoid low blood glucose (hypoglycaemia). If you have previously taken vildagliptin but had to stop taking it because of liver disease, you should not take this medicine. Diabetic skin lesions are a common complication of diabetes. You are advised to follow the recommendations for skin and foot care that you are given by your doctor or nurse. You are also advised to pay particular attention to new onset of blisters or ulcers while taking VIL GM. Should these occur, you should promptly consult your doctor. If you need to have major surgery you must stop taking VIL GM during and for some time after the procedure. Your doctor will decide when you must stop and when to restart your treatment with VIL GM. A test to determine your liver function will be performed before the start of VIL GM treatment, at three-month intervals for the first year and periodically thereafter. This is so that signs of increased liver enzymes can be detected as early as possible. During treatment with VIL GM, your doctor will check your kidney function at least once a year or more frequently if you are elderly and/or have worsening renal function. Your doctor will test your blood and urine for sugar regularly
Children and adolescents
The use of VIL GM in children and adolescents up to 18 years of age is not recommended.
Other medicines and VIL GM
If you need to have an injection of a contrast medium that contains iodine into your bloodstream, for example in the context of an X-ray or scan, you must stop taking VIL GM before or at the time of the injection. Your doctor will decide when you must stop and when to restart your treatment with VIL GM.
Tell your doctor if you are taking, have recently taken or might take any other medicines. You may need more frequent blood glucose and kidney function tests, or your doctor may need to adjust the dosage of VIL GM. It is especially important to mention the following:
- glucocorticoids generally used to treat inflammation
- beta-2 agonists generally used to treat respiratory disorders
- other medicines used to treat diabetes
- medicines which increase urine production (diuretics)
- medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as ibuprofen and celecoxib)
- certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists)
- certain medicines affecting the thyroid, or
- certain medicines affecting the nervous system.
VIL GM with alcohol
Avoid excessive alcohol intake while taking VIL GM since this may increase the risk of lactic acidosis (please see section “Warnings and precautions”).
Pregnancy and breast-feeding
If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor for advice before taking this medicine. Your doctor will discuss with you the potential risk of taking VIL GM during pregnancy. Do not use VIL GM if you are pregnant or breast-feeding (see also “Do not take VIL GM”). Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
If you feel dizzy while taking VIL GM, do not drive or use any tools or machines.
3. How to Use Vil Gm
The amount of VIL GM that people have to take varies depending on their condition. Your doctor will tell you exactly the dose of VIL GM to take.
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure. The recommended dose is one film-coated tablet of either 50 mg/5000 mg or 50 mg/1000 mg taken twice a day If you have reduced kidney function, your doctor may prescribe a lower dose. Also if you are taking an anti-diabetic medicine known as a sulphonylurea your doctor may prescribe a lower dose. Your doctor may prescribe this medicine alone or with certain other medicines that lower the level of sugar in your blood. When and how to take VIL GM
- Swallow the tablets whole with a glass of water,
- Take one tablet in the morning and the other in the evening with or just after food. Taking the tablet just after food will lower the risk of an upset stomach.
Continue to follow any advice about diet that your doctor has given you. In particular, if you are following a diabetic weight control diet, continue with this while you are taking VIL GM.
If you take more VIL GM than you should
If you take too many VIL GM tablets, or if someone else takes your tablets, talk to a doctor or pharmacist immediately. Medical attention may be necessary. If you have to go to a doctor or hospital, take the pack and this leaflet with you.
If you forget to take VIL GM
If you forget to take a tablet, take it with your next meal unless you are due to take one then anyway.
Do not take a double dose (two tablets at once) to make up for a forgotten tablet.
If you stop taking VIL GM
Continue to take this medicine as long as your doctor prescribes it so that it can continue to control your blood sugar. Do not stop taking VIL GM unless your doctor tells you to. If you have any questions about how long to take this medicine, talk to your doctor. If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
You should stop taking VIL GM and see your doctor immediately if you experience the following side effects:
- Lactic acidosis (very rare: may affect up to 1 user in 10,000): VIL GM may cause a very rare, but very serious side effect called lactic acidosis (see section “Warnings and precautions”). If this happens you must stop taking VIL GM and contact a doctor or the nearest hospital immediately, as lactic acidosis may lead to coma.
- Angioedema (rare: may affect up to 1 in 1,000 people): Symptoms include swollen face, tongue or throat, difficulty swallowing, difficulty breathing, sudden onset of rash or hives, which may indicate a reaction called “angioedema”.
- Liver disease (hepatitis) (rare): Symptoms include yellow skin and eyes, nausea, loss of appetite or dark-coloured urine, which may indicate liver disease (hepatitis).
- Inflammation of the pancreas (pancreatitis) (frequency not known): Symptoms include severe and persistent pain in the abdomen (stomach area), which might reach through to your back, as well as nausea and vomiting.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top right end of the home page. By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to Store Vil Gm
Store protect from moisture, at a temperature not exceeding 30°C.
Keep out of reach of children.
Do not use this medicine after the expiry date which is stated on the blister and the carton after “EXP”. The expiry date refers to the last day of that month.
6. Contents of the Pack and Other Information
What VIL GM contains
Vil GM 500
Each film coated tablet contains
Vildagliptin 50 mg
Metformin hydrochloride 500 mg
Excipients q.s.
Colours : Yellow Oxide of Iron and Titanium Dioxide IP
Vil GM 1000
Each film coated tablet contains Vildagliptin 50 mg
Metformin hydrochloride 1000 mg
Excipients q.s.
Colours : Yellow Oxide of Iron and Titanium Dioxide IP
© Zuventus Healthcare Ltd., 2020. All rights reserved.
For More Information About This Product
Dapafor V 10/100 mg Tablets
1.0 Generic name
Vildagliptin (As Sustained Release 100 mg) and Dapagliflozin 5 / 10 mg
2.0 Qualitative and quantitative formula of active ingredients
Dapafor-V 5/100 mg
Each bilayered tablet contains :
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin 5 mg
Vildagliptin IP 100 mg
(As Sustained Release)
Excipients q.s.
Colour : Ferric Oxide Red USP-NF
Dapafor-V 10/100 mg
Each bilayered tablet contains :
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin 10 mg
Vildagliptin IP 100 mg
(As Sustained Release)
Colour : Ferric Oxide Yellow USP-NF
Excipients q.s.
3.0 Dosage form and strength
Bilayer tablets, 5 mg / 100 mg and 10 mg / 100 mg
4.0 Clinical particulars
4.1 Therapeutic indication
Dapagliflozin and Vildagliptin Sustained Release tablets are indicated for the treatment of type 2 diabetes mellitus who are inadequately controlled on metformin monotherapy.
4.2 Posology and method of administration
Posology
The recommended dose is one tablet daily. Each tablet contains a fixed dose of dapagliflozin and Vildagliptin (As sustained Release)
Special populations
Renal impairment
No dose adjustment is required in patients with mild renal impairment (creatinine clearance ≥ 50 ml/min). In patients with moderate or severe renal impairment or with end-stage renal disease (ESRD), Dapagliflozin and Vildagliptin SR tablet is not recommended.
Hepatic impairment
This medicinal product must not be used in patients with hepatic impairment
Elderly (≥ 65 years)
This medicinal product should be used with caution as age increases.
Paediatric population
The safety and efficacy of Dapagliflozin and Vildagliptin SR tablets have not yet been established. No data are available.
Method of administration
Dapagliflozin and Vildagliptin SR tablets should be given once daily with meals to reduce the gastrointestinal adverse reactions.
4.3 Contraindications
Dapagliflozin and Vildagliptin SR tablets is contraindicated in patients with: hypersensitivity to the active substances or to any of the excipients used in the manufacturing of the finished product, any type of acute metabolic acidosis, diabetic pre-coma, severe renal failure, dehydration, severe infection, shock, cardiac or respiratory failure, acute alcohol intoxication and alcoholism.
4.4 Special warnings and precautions for use
Dapagliflozin
Volume depletion : Before initiating Dapagliflozin, assess volume status and renal function in the elderly, patients with renal impairment or low systolic blood pressure, and in patients on diuretics. Monitor for signs and symptoms during therapy.
Ketoacidosis in Patients with Diabetes Mellitus : Assess patients who present with signs and symptoms of metabolic acidosis for ketoacidosis regardless of blood glucose level. If suspected, discontinue Dapagliflozin, evaluate and treat promptly. Before initiating Dapagliflozin, consider risk factors for ketoacidosis. Patients on Dapagliflozin may require monitoring and temporary discontinuation of therapy in clinical situations known to predispose to ketoacidosis.
Urosepsis and Pyelonephritis : Evaluate for signs and symptoms of urinary tract infections and treat promptly, if indicated.
Hypoglycemia : Consider a lower dose of insulin or the insulin secretagogue to reduce the risk of hypoglycemia when used in combination with Dapagliflozin.
Necrotizing Fasciitis of the Perineum (Fournier's Gangrene) : Serious, life-threatening cases have occurred in patients with diabetes, both females and males. Assess patients presenting with pain or tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise. If suspected, institute prompt treatment.
Genital Mycotic Infections : Monitor and treat if indicated.
Elderly (≥ 65 years)
Elderly patients may be at a greater risk for volume depletion and are more likely to be treated with diuretics. Elderly patients are more likely to have impaired renal function, and/or to be treated with anti-hypertensive medicinal products that may cause changes in renal function such as angiotensin-converting enzyme inhibitors (ACE-I) and angiotensin II type 1 receptor blockers (ARB). The same recommendations for renal function apply to elderly patients as to all patients.
Lower limb amputations
An increase in cases of lower limb amputation (primarily of the toe) has been observed in long-term, clinical studies in type 2 diabetes mellitus with SGLT2 inhibitors. It is unknown whether this constitutes a class effect. It is important to counsel patients with diabetes on routine preventative foot care.
Vildagliptin
General
Vildagliptin is not a substitute for insulin in insulin-requiring patients. Vildagliptin should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis.
Renal impairment
There is limited experience in patients with ESRD on haemodialysis. Therefore Vildagliptin should be used with caution in these patients (see also sections 4.2, 5.1 and 5.2).
Hepatic impairment
Vildagliptin should not be used in patients with hepatic impairment, including patients with pre-treatment ALT or AST > 3x ULN (see also sections 4.2 and 5.2).
Liver enzyme monitoring
Rare cases of hepatic dysfunction (including hepatitis) have been reported. In these cases, the patients were generally asymptomatic without clinical sequelae and liver function test results returned to normal after discontinuation of treatment. Liver function tests should be performed prior to the initiation of treatment with Vildagliptin in order to know the patient's baseline value. Liver function should be monitored during treatment with Vildagliptin at three-month intervals during the first year and periodically thereafter. Patients who develop increased transaminase levels should be monitored with a second liver function evaluation to confirm the finding and be followed thereafter with frequent liver function tests until the abnormality(ies) return(s) to normal. Should an increase in AST or ALT of 3x ULN or greater persist, withdrawal of Vildagliptin therapy is recommended. Patients who develop jaundice or other signs suggestive of liver dysfunction should discontinue Vildagliptin. Following withdrawal of treatment with Vildagliptin and LFT normalisation, treatment with Vildagliptin should not be reinitiated.
Cardiac failure
A clinical trial of vildagliptin in patients with New York Heart Association (NYHA) functional class I-III showed that treatment with vildagliptin was not associated with a change in left-ventricular function or worsening of pre-existing congestive heart failure (CHF) versus placebo. Clinical experience in patients with NYHA functional class III treated with vildagliptin is still limited and results are inconclusive (see section 5.1). There is no experience of vildagliptin use in clinical trials in patients with NYHA functional class IV and therefore use is not recommended in these patients.
Skin disorders
Skin lesions, including blistering and ulceration have been reported in extremities of monkeys in non-clinical toxicology studies (see section 5.3). Although skin lesions were not observed at an increased incidence in clinical trials, there was limited experience in patients with diabetic skin complications. Furthermore, there have been post-marketing reports of bullous and exfoliative skin lesions. Therefore, in keeping with routine care of the diabetic patient, monitoring for skin disorders, such as blistering or ulceration, is recommended.
Acute pancreatitis
Use of vildagliptin has been associated with a risk of developing acute pancreatitis. Patients should be informed of the characteristic symptom of acute pancreatitis.
If pancreatitis is suspected, vildagliptin should be discontinued; if acute pancreatitis is confirmed, vildagliptin should not be restarted. Caution should be exercised in patients with a history of acute pancreatitis.
Hypoglycaemia
Sulphonylureas are known to cause hypoglycaemia. Patients receiving vildagliptin in combination with a sulphonylurea may be at risk for hypoglycaemia. Therefore, a lower dose of sulphonylurea may be considered to reduce the risk of hypoglycaemia.
Arthralgia :
We identified cases of severe joint pain associated with the use of DPP-4 inhibitors. Patients started having symptoms from 1 day to years after they started taking a DPP-4 inhibitor. After the patients discontinued the DPP-4 inhibitor medicine, their symptoms were relieved, usually in less than a month. Some patients developed severe joint pain again when they restarted the same medicine or another DPP-4 inhibitor. In such case, Patients should not stop taking their DPP-4 inhibitor medicine, but should contact their health care professional right away if they experience severe and persistent joint pain.
4.5 Interaction with other medicinal products and other forms of interaction
No interaction studies have been performed for Dapagliflozin and Vildaglitpin SR tablets. The following statements reflect the information available on the individual active substances.
Dapagliflozin
Pharmacodynamic interactions
Diuretics
Dapagliflozin may add to the diuretic effect of thiazide and loop diuretics and may increase the risk of dehydration and hypotension.
Insulin and insulin secretagogues
Insulin and insulin secretagogues, such as sulphonylureas, cause hypoglycaemia. Therefore, a lower dose of insulin or an insulin secretagogue may be required to reduce the risk of hypoglycaemia when used in combination with Dapagliflozin in patients with type 2 diabetes mellitus. In patients with type 1 diabetes mellitus and a known risk of frequent or severe hypoglycaemia, it may be necessary to reduce the insulin dose at the time of initiating treatment with Dapagliflozin to decrease the risk of hypoglycaemia. When needed, insulin dose reduction should be done cautiously to avoid ketosis and DKA.
Pharmacokinetic interactions
The metabolism of Dapagliflozin is primarily via glucuronide conjugation mediated by UDP glucuronosyltransferase 1A9 (UGT1A9). In in vitro studies, Dapagliflozin neither inhibited cytochrome P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, nor induced CYP1A2, CYP2B6 or CYP3A4. Therefore, Dapagliflozin is not expected to alter the metabolic clearance of co administered medicinal products that are metabolised by these enzymes.
Vildagliptin
Vildagliptin has a low potential for interactions with co-administered medicinal products. Since vildagliptin is not a cytochrome P (CYP) 450 enzyme substrate and does not inhibit or induce CYP 450 enzymes, it is not likely to interact with active substances that are substrates, inhibitors or inducers of these enzymes.
Digoxin (Pgp substrate), warfarin (CYP2C9 substrate)
Clinical studies performed with healthy subjects have shown no clinically relevant pharmacokinetic interactions. However, this has not been established in the target population.
Combination with amlodipine, ramipril, valsartan or simvastatin
Drug-drug interaction studies in healthy subjects were conducted with amlodipine, ramipril, valsartan and simvastatin. In these studies, no clinically relevant pharmacokinetic interactions were observed after co-administration with vildagliptin.
Combination with ACE-inhibitors
There may be an increased risk of angioedema in patients concomitantly taking ACE-inhibitors. As with other oral antidiabetic medicinal products the hypoglycaemic effect of vildagliptin may be reduced by certain active substances, including thiazides, corticosteroids, thyroid products and sympathomimetics.
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no data from the use of Dapagliflozin and Vildagliptin SR tablets or Dapagliflozin in pregnant women. There are no adequate data from the use of vildagliptin in pregnant women. Studies in animals have shown reproductive toxicity at high doses (see section 5.3). The potential risk for humans is unknown. Due to lack of human data, Vildagliptin should not be used during pregnancy.
Breast-feeding
It is unknown whether this medicinal product or dapagliflozin (and / or its metabolites) are excreted in human milk. Available pharmacodynamic / toxicological data in animals have shown excretion of dapagliflozin/metabolites in milk, as well as pharmacologically-mediated effects in nursing offspring. It is unknown whether vildagliptin is excreted in human milk. Animal studies have shown excretion of vildagliptin in milk. Vildagliptin should not be used during breast-feeding.
Fertility
The effect of this medicinal product or Dapagliflozin & Vildagliptin on fertility in humans has not been studied.
4.7 Effects on ability to drive and use machines
Dapagliflozin and Vildagliptin SR tablets have no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Dapagliflozin and Vildagliptin SR tablets have been demonstrated to be bioequivalent with co administered Dapagliflozin and Vildagliptin. There have been no therapeutic clinical trials conducted with Dapagliflozin and Vildagliptin SR tablets. As per reported evidences, following adverse reactions have been identified in the placebo-controlled clinical studies. None were found to be dose-related. Adverse reactions listed below are classified according to frequency and system organ class (SOC). Frequency categories are defined according to the following convention : very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), and not known (cannot be estimated from the available data).

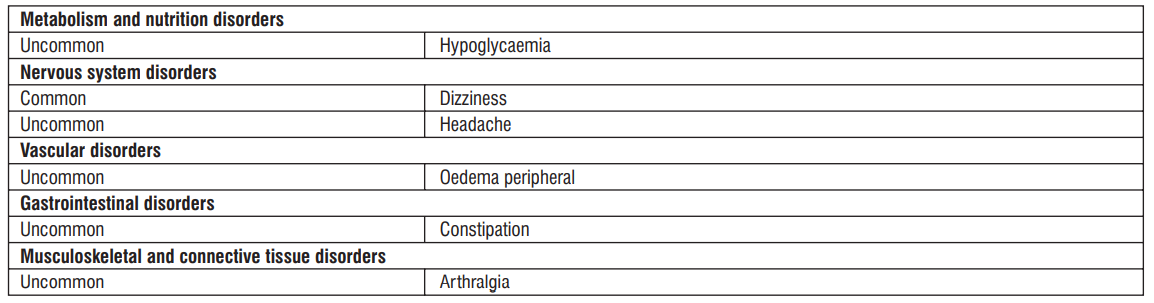
Dapagliflozin
Very common : Hypoglycaemia (when used with SU or insulin)
Common : Vulvovaginitis, balanitis and related genital infections, Urinary tract infection
- Dizziness
- Rash
- Back pain
- Dysuria, Polyuria
- Dyslipidaemia
- Haematocrit increased, Creatinine renal clearance decreased during initial treatment
Uncommon : Fungal infection
- Volume depletion, Thirst
- Constipation, Dry mouth
- Nocturia
- Vulvovaginal pruritus, Pruritus genital
- Blood creatinine increased during initial treatment, Blood urea increased
- Weight decreased
Rare : Diabetic ketoacidosis (when used in type 2 diabetes mellitus)
Very rare : Necrotising fasciitis of the perineum (Fournier's gangrene)
- Angioedema
Vildagliptin
Adverse reactions reported in patients who received vildagliptin as monotherapy
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com
4.9 Overdose
Dapagliflozin
Dapagliflozin did not show any toxicity in healthy subjects at single oral doses up to 500 mg (50 times the maximum recommended human dose).
Vildagliptin
Information on the likely symptoms of overdose was taken from a rising dose tolerability study in healthy subjects given Vildagliptin for 10 days. At 400 mg, there were three cases of muscle pain, and individual cases of mild and transient paraesthesia, fever, oedema and a transient increase in lipase levels. At 600 mg, one subject experienced oedema of the feet and hands, and increases in creatine phosphokinase (CPK), aspartate aminotransferase (AST), C-reactive protein (CRP) and myoglobin levels. Three other subjects experienced oedema of the feet, with paraesthesia in two cases. All symptoms and laboratory abnormalities resolved without treatment after discontinuation of the study medicinal product.
5.0 Pharmacological properties
5.1 Pharmacodynamic properties
Pharmacotherapeutic group : Drugs used in diabetes, Combinations of oral blood glucose-lowering drugs.
Mechanism of action
Dapagliflozin is a reversible inhibitor of sodium-glucose co-transporter 2 (SGLT2) that improves glycemic control in patients with type 2 diabetes mellitus by reducing renal glucose reabsorption leading to urinary excretion of excess glucose (glucuresis). SGLT2 is selectively expressed in the kidney. SGLT2 is the predominant transporter responsible for reabsorption of glucose from the glomerular filtrate back into the circulation. Dapagliflozin improves both fasting and post-prandial plasma glucose levels by reducing renal glucose reabsorption leading to urinary excretion of excess glucose. The amount of glucose removed by the kidney through this mechanism is dependent upon the blood glucose concentration and GFR. Dapagliflozin does not impair normal endogenous glucose production in response to hypoglycemia. Dapagliflozin acts independently of insulin secretion and insulin action.
Urinary glucose excretion (glucuresis) induced by Dapagliflozin is associated with caloric loss and reduction in weight. Inhibition of glucose and sodium co-transport by Dapagliflozin is also associated with mild diuresis and transient natriuresis. The effect of vildagliptin layer results in a rapid and complete inhibition of DPP-4 activity, resulting in increased fasting and postprandial endogenous levels of the incretin hormones GLP-1 (glucagon-like peptide 1) and GIP (glucose-dependent insulinotropic polypeptide).
Vildagliptin
The administration of vildagliptin results in a rapid and complete inhibition of DPP-4 activity, resulting in increased fasting and postprandial endogenous levels of the incretin hormones GLP-1 (glucagon-like peptide 1) and GIP (glucose-dependent insulinotropic polypeptide).
Pharmacodynamic effects
Dapagliflozin
Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in subjects with type 2diabetes mellitus following the administration of dapagliflozin.
Vildagliptin
By increasing the endogenous levels of these incretin hormones, vildagliptin enhances the sensitivity of beta cells to glucose, resulting in improved glucosedependent insulin secretion. Treatment with vildagliptin sustained release tablets 100 mg daily in patients with type 2 diabetes significantly improved markers of beta cell function including HOMA-β (Homeostasis Model Assessment-β), proinsulin to insulin ratio and measures of beta cell responsiveness from the frequentlysampled meal tolerance test. In non-diabetic (normal glycaemic) individuals, vildagliptin does not stimulate insulin secretion or reduce glucose levels. By increasing endogenous GLP-1 levels, vildagliptin also enhances the sensitivity of alpha cells to glucose, resulting in more glucose-appropriate glucagon secretion.
The enhanced increase in the insulin/glucagon ratio during hyperglycaemia due to increased incretin hormone levels results in a decrease in fasting and postprandial hepatic glucose production, leading to reduced glycaemia. The known effect of increased GLP-1 levels delaying gastric emptying is not observed with vildagliptin treatment.
5.2 Pharmacokinetic properties
Dapagliflozin
Absorption : Dapagliflozin was rapidly and well absorbed after oral administration and can be administered with or without food. Geometric mean steady-state Dapagliflozin Cmax and AUCτ values following once daily 10 mg doses of Dapagliflozin were 158 ng/mL and 628 ng.h/mL, respectively. Maximum Dapagliflozin plasma concentrations (Cmax) were usually attained within 2 hours after administration in the fasted state. The Cmax and AUC values increased proportionally to the increment in Dapagliflozin dose. The absolute oral bioavailability of Dapagliflozin following the administration of a 10 mg dose is 78%. Food had relatively modest effects on the pharmacokinetics of Dapagliflozin in healthy subjects. Administration with a high-fat meal decreased Dapagliflozin Cmax by up to 50% and prolonged Tmax by approximately 1 hour, but did not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful. Distribution : Dapagliflozin is approximately 91% protein bound. Protein binding was not altered in various disease states (e.g. renal or hepatic impairment). Metabolism : Dapagliflozin is a C-linked glucoside, meaning the aglycone component is attached to glucose by a carbon-carbon bond, thereby conferring stability against glucosidase enzymes. The mean plasma terminal half-life (t1/2) for Dapagliflozin was 12.9 hours following a single oral dose of Dapagliflozin 10 mg to healthy subjects. Dapagliflozin is extensively metabolized, primarily to yield Dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O-glucuronide accounted for 61% of a 50 mg [14C]-Dapagliflozin dose and was the predominant drug-related component in human plasma, accounting for 42% (based on AUC [0-12 h]) of total plasma radioactivity, similar to the 39% contribution by parent drug. Based on AUC, no other metabolite accounted for >5% of the total plasma radioactivity at any time point measured. Dapagliflozin 3-O-glucuronide or other metabolites do not contribute to the glucose-lowering effects. The formation of Dapagliflozin 3-O-glucuronide is mediated by UGT1A9, an enzyme present in the liver and kidney, and CYP-mediated metabolism was a minor clearance pathway in humans. Excretion : Dapagliflozin and related metabolites are primarily eliminated via urinary excretion, of which less than 2% is unchanged Dapagliflozin. After administration of 50 mg [14C]-Dapagliflozin dose, 96% was recovered, 75% in urine and 21% in feces. In feces, approximately 15% of the dose was excreted as parent drug
Vildagliptin
Absorption
Following oral administration in the fasting state, Vildagliptin is rapidly absorbed, with peak plasma concentrations observed at 1.7 hours. Food slightly delays the time to peak plasma concentration to 2.5 hours, but does not alter the overall exposure (AUC). Administration of Vildagliptin with food resulted in a decreased Cmax (19%). However, the magnitude of change is not clinically significant, so that Vildagliptin can be given with or without food. The absolute bioavailability is 85%.
Distribution
The plasma protein binding of Vildagliptin is low (9.3%) and Vildagliptin distributes equally between plasma and red blood cells. The mean volume of distribution of Vildagliptin at steady-state after intravenous administration (Vss) is 71 litres, suggesting extravascular distribution.
Biotransformation
Metabolism is the major elimination pathway for Vildagliptin in humans, accounting for 69% of the dose. The major metabolite (LAY 151) is pharmacologically inactive and is the hydrolysis product of the cyano moiety, accounting for 57% of the dose, followed by the glucuronide (BQS867) and the amide hydrolysis products (4% of dose). In vitro data in human kidney microsomes suggest that the kidney may be one of the major organs contributing to the hydrolysis of Vildagliptin to its major inactive metabolite, LAY151. DPP-4 contributes partially to the hydrolysis of Vildagliptin based on an in vivo study using DPP-4 deficient rats. Vildagliptin is not metabolised by CYP 450 enzymes to any quantifiable extent. Accordingly, the metabolic clearance of Vildagliptin is not anticipated to be affected by co-medications that are CYP 450 inhibitors and/or inducers. In vitro studies demonstrated that Vildagliptin does not inhibit/induce CYP 450 enzymes. Therefore, Vildagliptin is not likely to affect metabolic clearance of co-medications metabolised by CYP 1A2, CYP 2C8, CYP 2C9, CYP 2C19, CYP 2D6, CYP 2E1 or CYP 3A4/5.
Elimination
Following oral administration of [14C] Vildagliptin, approximately 85% of the dose was excreted into the urine and 15% of the dose is recovered in the faeces. Renal excretion of the unchanged Vildagliptin accounted for 23% of the dose after oral administration. After intravenous administration to healthy subjects, the total plasma and renal clearances of Vildagliptin are 41 and 13 l/h, respectively. The mean elimination half-life after intravenous administration is approximately 2 hours. The elimination half-life after oral administration is approximately 3 hours.
5.3 Preclinical safety data
Preclinical Pharmacology
Dapagliflozin :
In vivo primary pharmacodynamic studies with Dapagliflozin were carried out in single-dose, dose ranging studies in non-diabetic and diabetic rats or mice in order to evaluate the potency, SGLT2-specificity and duration of action in stimulating urinary glucose excretion, and to describe the secondary consequences of urinary glucose excretion, such as changes in urine volume or blood or plasma glucose effects. Subsequently a multiple-dose study was carried out to evaluate the ability of Dapagliflozin to have sustained effects on urinary glucose excretion, urine volume, and fasting plasma glucose in diabetic rats over a two-week dosing period. Dapagliflozin increased renal glucose excretion in (healthy, non-diabetic) experimental animals. This was accompanied, by osmotic diuresis as measured by increased urine flow. An oral glucose tolerance test was also performed showing that Dapagliflozin was able to significantly reduce glucose area under the curve (AUC), compared to vehicle treatment. A study in knock-out mice lacking the gene for SGLT2 revealed that SGLT2 is indeed the main target for Dapagliflozin at least at lower doses. This study also demonstrated the reversibility of Dapagliflozin's action towards SGLT2. Vildagliptin is a selective and potent inhibitor of DPP-4. The IC50 value for inhibition of human DPP-4 is about 3 nM and similar activity was observed with the rat enzyme, demonstrating the lack of species selectivity. Vildagliptin showed some activity at the related enzymes DPP-8 and DPP-9 (Ki values of 506 nM and 65 nM respectively). Although these values are 253 and 32 times higher than the Ki for DPP-4, activity at Cmax in humans (2.3 μM) is likely. No assays exist allowing evaluation of DPP-8 / DPP-9 inhibition in vivo. The possibility of activity at one or both of these targets is considered a safety concern in relation to the occurrence of skin lesions in monkeys. No, or minimal, inhibition was seen with other related enzymes. In vivo pharmacodynamic studies were performed in rats and monkeys. These studies demonstrated the in vivo inhibition of DPP-4 and increased plasma levels of GLP-1. Studies in diabetic rats and in insulin-resistant monkeys demonstrated a glucose-lowering effect of Vildagliptin. Chronic effects of Vildagliptin were studied in pre-diabetic and insulin-treated diabetic monkeys. Beneficial effects were observed on HbA1c, fasting insulin, fibrinogen and PAI-1.
6.0 Pharmaceutical particulars
6.1 Incompatibilities
Not applicable
6.2 Special precautions for storage
This medicinal product does not require any special storage conditions.
6.3 Nature and contents of container
Alu-Alu blister. 10 bilayer tablets in non-perforated blisters.
6.4 Special precautions for disposal and other handling
Any unused medicinal product or waste material should be disposed of in accordance with local requirements
7.0 Shelf-life
Refer on the pack
8.0 Packaging information
A blister strip of 10 tablets
9.0 Storage condition
Store below 30°C. Protect from light & moisture.
Keep out of reach of children.
12.0 Date of issue
22 June 2022

About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet:
- What Dapafor-V is and what it is used for
- What you need to know before you take Dapafor-V
- How to take Dapafor-V
- Possible side effects
- How to store Dapafor-V
- Contents of the pack and other information
1. What Dapafor-V is and what it is used for
Dapafor-V contains the active substances vildagliptin and dapagliflozin. Each belongs to a group of medicines called “oral anti-diabetics”. These medicines are taken by mouth for diabetes.
Dapafor-V is used for a type of diabetes called “type 2 diabetes mellitus” in adult patients (aged 18 years and older). If you have type 2 diabetes, your pancreas does not make enough insulin or your body is not able to use the insulin it produces properly. This leads to a high level of sugar in your blood. The two active substances in Dapafor-V work in different ways to help control the level of sugar in your blood and remove excess sugar from your body via your urine.
Dapafor-V is used to treat type 2 diabetes when:
- vildagliptin or dapagliflozin alone together with metformin and/or sulphonylurea cannot control your diabetes.
- you are already being treated with vildagliptin and dapagliflozin as single tablets. Your doctor may ask you to switch to this medicine.
It is important to continue to follow the advice on diet and exercise given to you by your doctor, pharmacist or nurse.
2. What you need to know before you take Dapafor-V
Do not take Dapafor-V:
- if you are allergic to vildagliptin, dapagliflozin or any of the other ingredients of this medicine (listed in section 6).
- if you have had a serious allergic reaction to any other similar medicines (for example DPP-4 inhibitors like sitagliptin, linagliptin, alogliptin, or SGLT2 inhibitors like canagliflozin, empagliflozin) that you take to control your blood sugar.
Do not take Dapafor-V if any of the above apply to you. If you are not sure, talk to your doctor, pharmacist, or nurse before taking this medicine.
Warnings and precautions
Talk to your doctor, pharmacist or nurse before taking Dapafor-V, and during treatment:
- if you have or have had a disease of the pancreas called pancreatitis. Possible signs of pancreatitis are listed in section 4.
- if you are on medicines to lower your blood pressure (anti-hypertensives) and have a history of low blood pressure (hypotension). For more information, see section “Other medicines and Dapafor-V” below.
- if you have very high levels of sugar in your blood which may make you dehydrated (lose too much body fluid). Possible signs of dehydration are listed at the top of section 4. Tell your doctor before you start taking Dapafor-V if you have any of these signs.
- if you have or develop nausea (feeling sick), vomiting or fever or if you are not able to eat or drink. These conditions can cause dehydration. Your doctor may ask you to stop taking Dapafor-V until you recover to prevent dehydration.
- if you have moderate or severe liver problem.
- if you experience rapid weight loss, feeling sick or being sick, stomach pain, excessive thirst, fast and deep breathing, confusion, unusual sleepiness or tiredness, a sweet smell to your breath, a sweet or metallic taste in your mouth, or a different odour to your urine or sweat, contact a doctor or the nearest hospital straight away. These symptoms could be a sign of “diabetic ketoacidosis” – a rare but serious, sometimes life-threatening problem you can get with diabetes because of increased levels of “ketone bodies” in your urine or blood, seen in tests. The risk of developing diabetic ketoacidosis may be increased with prolonged fasting, excessive alcohol consumption, dehydration, sudden reductions in insulin dose, or a higher need of insulin due to major surgery or serious illness.
- if you have “type 1 diabetes” your body does not produce any insulin. Dapafor-V should not be used to treat this condition.
- if you have or have had a serious hypersensitivity (allergic) reaction or is suspected. Signs of a serious allergic reaction are listed in section 4.
- if you often get infections of the urinary tract.
- if you have a history of serious heart disease.
- if you suffer from heart failure or you have other risk factors for developing heart failure such as problems with your kidneys. Your doctor will advise you of the signs and symptoms of heart failure. Symptoms can include, but are not limited to, increasing shortness of breath, rapid increase in weight and swelling of the feet (pedal oedema). You should call your doctor, pharmacist or nurse immediately if you experience any of these symptoms.
- if you have severe joint pain.
- if your body’s ability to fight infections is reduced, for example if you have a disease like AIDS or have undergone an organ transplant.
- if you are taking a medicine to lower your blood sugar, such as sulphonylureas (see “Other medicines and Dapafor-V”).
If any of the above apply to you (or you are not sure), talk to your doctor, pharmacist or nurse before taking Dapafor-V.
Diabetic skin lesions (skin damage such as sores or ulcers) are a common complication of diabetes. Rash has been seen with both vildagliptin and dapagliflozin when given separately (see section 4). You are advised to follow the recommendations for skin care that you are given by your doctor or nurse. Contact your doctor if you encounter blistering of the skin, as it may be a sign for a condition called bullous pemphigoid. Your doctor may ask you to stop Dapafor-V.
Like for all diabetic patients it is important to check your feet regularly and adhere to any other advice regarding foot care given by your health care professional.
Talk to your doctor immediately if you develop a combination of symptoms of pain, tenderness, redness, or swelling of the genitals or the area between the genitals and the anus with fever or feeling generally unwell. These symptoms could be a sign of a rare but serious or even life-threatening infection, called necrotising fasciitis of the perineum or Fournier’s gangrene which destroys the tissue under the skin. Fournier’s gangrene has to be treated immediately.
Kidney function
Your kidneys should be checked before you start taking Dapafor-V. During treatment with this medicine, your doctor will check your kidney function once a year or more frequently if you have worsening kidney function.
Urine tests
Because of how Dapafor-V works, your urine will test positive for sugar while you are on this medicine.
Children and adolescents
Dapafor-V is not recommended for children and adolescents under 18 years of age, because it has not been studied in these patients.
Other medicines and Dapafor-V
Tell your doctor, pharmacist or nurse if you are taking, have recently taken or might take any other medicines.
Especially tell your doctor:
- if you are taking a medicine used to increase the amount of water you pass out of the body (diuretic). Your doctor may ask you to stop taking Dapafor-V. Possible signs of losing too much fluid from your body are listed at the top of section 4.
- if you are taking another medicine that lowers the amount of sugar in your blood such as a sulphonylurea (for example glimepiride). Your doctor may want to lower the dose of this other medicine, to prevent you from getting low blood sugar levels (hypoglycaemia).
- if you are using medicines containing any of the following active substances, that might have an effect on the breakdown of Dapafor-V in your body. Your doctor may ask you to check your blood sugar levels more often while taking these medicines.
- Carbamazepine, phenobarbital or phenytoin. These may be used to control fits (seizures) or chronic pain.
- Dexamethasone – a steroid medicine. This may be used to treat inflammation in different body parts and organs.
- Rifampicin. This is an antibiotic used to treat infections such as tuberculosis. Ketoconazole. This may be used to treat fungal infections.
- Diltiazem. This is a medicine used to treat angina (chest pain) and lower blood pressure. If any of the above apply to you (or if you are not sure), talk to your doctor before taking Dapafor-V.
Pregnancy and breast-feeding
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine. Dapafor-V is not recommended during pregnancy and your doctor will ask you to stop taking this medicine if you become pregnant. Talk to your doctor about the best way to control your blood sugar while you are pregnant.
You should not use Dapafor-V if you are breast-feeding. It is not known if this medicine passes into human breast milk. Talk to your doctor if you would like to or are breast-feeding before taking this medicine.
Driving and using machines
Dapafor-V is not expected to affect you being able to drive a car or use any tools or machines. If you feel dizzy while taking this medicine, do not drive or use any tools or machines. Taking this medicine together with another medicine that lowers your blood sugar, such as a sulphonylurea, can cause too low blood sugar levels (hypoglycaemia). This may cause symptoms such as shaking, sweating and change in vision, and may affect your ability to drive and use machines.
Dapafor-V contains lactose
Dapafor-V contains lactose (milk sugar). If you have been told by your doctor that you have an intolerance to some sugars, contact your doctor before taking this medicine.
Dapafor-V contains sodium
Dapafor-V contains less than 1 mmol sodium (23 mg) per dose, that is to say essentially ‘sodiumfree’.
3. How to take Dapafor-V
Always take this medicine exactly as your doctor has told you. Check with your doctor, pharmacist or nurse if you are not sure.
How much to take
- The recommended dose is one tablet a day.
Taking this medicine
- Swallow the tablet whole with half a glass of water.
- You can take your tablet with or without food.
- You can take the tablet at any time of the day. However, try to take it at the same time each day. This will help you to remember to take it.
Your doctor may prescribe other medicines to lower the amount of sugar in your blood. Remember to take other medicine(s) as your doctor has told you. This will help get the best results for your health.
Diet and exercise
To control your diabetes, you still need to keep to diet and exercise, even when you are taking this medicine. So it is important to keep following the advice about diet and exercise from your doctor, pharmacist or nurse. In particular, if you are following a diabetic weight control diet, continue to follow it while you are taking Dapafor-V.
If you take more Dapafor-V than you should
If you take more Dapafor-V tablets than you should, talk to a doctor or go to a hospital straight away. Take the medicine pack with you.
If you forget to take Dapafor-V
What to do if you forget to take a tablet.
- If it is less than 12 hours since you should have taken your dose, take a dose of Dapafor-V as soon as you remember. Then take your next dose at the usual time.
- If it is more than 12 hours since you should have taken your dose, skip the missed dose. Then take your next dose at the usual time.
- Do not take a double dose of Dapafor-V to make up for a forgotten dose.
If you stop taking Dapafor-V
Do not stop taking Dapafor-V without talking to your doctor first. Your blood sugar may increase without this medicine.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Stop taking Dapafor-V and see a doctor straight away if you notice any of the following serious side effects:
Symptoms of a serious allergic reaction (anaphylactic reaction, angioedema) seen rarely, (may affect up to 1 in 1 000 people), which may include:
- rash,
- raised red patches on your skin (hives),
- swelling of the face,lips, tongue, and throat that may cause difficulty in breathing or
- swallowing.
Your doctor may prescribe a medicine to treat your allergic reaction and a different medicine for your diabetes.
Pancreatitis, seen uncommonly (may affect up to 1 in 100 people): severe and persistent pain in the abdomen (stomach area) which might reach through to your back, as well as nausea and vomiting, as it could be a sign of an inflamed pancreas.
Dehydration, (loss of too much fluid from your body), seen uncommonly. These are signs of dehydration:
- very dry or sticky mouth, feeling very thirsty,
- feeling very sleepy or tired, - passing little or no water (urine), - fast heart beat.
Urinary tract infection, seen commonly (may affect up to 1 in 10 people). These are signs of a severe infection of the urinary tract:
- fever and/or chills, burning sensation when passing water (urinating),
- pain in your back or side.
Although uncommon, if you see blood in your urine, tell your doctor immediately.
Low blood sugar levels (hypoglycaemia), seen very commonly (may affect more than 1 in 10 people) if used with other diabetes medicines known to cause hypoglycaemia.
These are the signs of low blood sugar:
shaking, sweating, feeling very anxious, fast heart beat, - feeling hungry, headache, change in vision, - a change in your mood or feeling confused.
Your doctor will tell you how to treat low blood sugar levels and what to do if you get any of the signs above.
Diabetic ketoacidosis, seen rarely.
These are the signs of diabetic ketoacidosis (see also section 2 Warnings and precautions): - increased levels of “ketone bodies” in your urine or blood,
- rapid weight loss,
- feeling sick or being sick,
- stomach pain,
- excessive thirst,
- fast and deep breathing,
- confusion,
- unusual sleepiness or tiredness,
- a sweet smell to your breath,
- a sweet or metallic taste in your mouth or a different odour to your urine or sweat.
This may occur regardless of blood glucose level. Your doctor may decide to temporarily or permanently stop your treatment with Dapafor-V.
Necrotising fasciitis of the perineum or Fournier’s gangrene, a serious soft tissue infection of the genitals or the area between the genitals and the anus, seen very rarely (may affect up to 1 in 10 000 people).
Stop taking Dapafor-V and see a doctor or nurse straight away, if you notice any of the serious side effects above.
Other side effects when taking Dapafor-V alone or in combination with metformin: Very common
- upper respiratory tract infection including: - infection of the upper chest or lungs,
- infection of the sinuses with a feeling of pain and fullness behind your cheeks and eyes (sinusitis),
- inflamed nose or throat (nasopharyngitis) (signs of this may include a cold or a sore throat).
Common
- genital infection (thrush) of your penis or vagina (signs may include irritation, itching, unusual discharge or odour)
- back pain
- passing more water (urine) than usual or needing to pass water more often
- changes in the amount of cholesterol or fats in your blood (shown in tests)
- increases in the amount of red blood cells in your blood (shown in tests)
- decreases in creatinine renal clearance (shown in tests) in the beginning of treatment
- dizziness
- tiredness
- severe joint pain (arthralgia)
- stomach ache and indigestion (dyspepsia)
- nausea
- diarrhoea
- inflamed stomach or gut usually caused by an infection (gastroenteritis)
- headache, muscle pain (myalgia)
- vomiting, inflammation of the stomach (gastritis)
- rash
Uncommon
- thirst
- constipation
- awakening from sleep at night to pass urine
- dry mouth
- weight decreased
- increases in creatinine (shown in laboratory blood tests) in the beginning of treatment
- increases in urea (shown in laboratory blood tests)
- skin rash that may include raised bumps, skin irritation, or unpleasant itchiness
- difficulties in getting or maintaining an erection (erectile dysfunction)
- fungal infection
- hypersensitivity reactions
- itching in the genital area (pruritus genital or vulvovaginal pruritus) or discomfort while urinating
Not known (frequency cannot be estimated from the available data)
- blistering of the skin (bullous pemphigoid)
Reporting of side effects
If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top of the home page. By reporting side effects, you can help provide more information on the safety of this medicine.
5. How to store Dapafor-V
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date, which is stated on the blister and carton after ‘EXP’. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions. Do not throw away any medicines via wastewater or household waste.
Ask your pharmacist how to throw away medicines you no longer use. These measures will help to protect the environment.
6. Contents of the pack and other information
What Dapafor-V contains
- The active substances are vildagliptin and dapagliflozin.
Dapafor-V 5/100 mg
Each bilayer tablet contains:
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin -5 mg
Vildagliptin IP -100 mg (Sustained Release)
Dapafor-V 10/100 mg
Each bilayer tablet contains:
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin- 10 mg
Vildagliptin IP 100 mg (Sustained Release)
What Dapafor-V looks like and contents of the pack
Dapafor-V 5/100 mg
Colour : Ferric Oxide Red USP-NF
Dapafor-V 10/100 mg
Colour : Ferric Oxide Yellow USP-NF
Pack contains 10 Blister Strips of 10 tablet each.
For More Information About This Product
Dapaformin 1000 Tablet
1.0 Generic name
Dapagliflozin & Metformin Hydrochloride Extended Release Tablets
2.0 Qualitative and quantitative composition
DapaFormin-500
Each film coated tablet contains :
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin 10 mg
Metformin Hydrochloride IP 500 mg
(Extended Release)
Excipients q.s Colours : Titanium Dioxide IP & Ferric Oxide Yellow USP-NF
DapaFormin-1000
Each film coated tablet contains :
Dapagliflozin Propanediol Monohydrate
equivalent to Dapagliflozin 10 mg
Metformin Hydrochloride IP 1000 mg
(Extended Release)
Excipients q.s
Colours : Titanium Dioxide IP & Ferric Oxide Yellow USP-NF
3.0 Dosage form and strength
Film coated tablet
4.0 Clinical particulars
4.1 Therapeutic indication
It is indicated as an adjunct to diet and exercise to improve glycaemic control in adults with type 2 diabetes mellitus when treatment with both Dapagliflozin and Metformin is appropriate.
4.2 Posology and method of administration
Dapagliflozin + Metformin ER tablet once daily in the morning with food or as directed by physician. Swallow Dapagliflozin Metformin ER tablets whole and never crush, cut, or chew
4.3 Contraindications
- Severe renal impairment (eGFR below 30 mL/min/1.73 m2 ), end stage renal disease or patients on dialysis
- History of a serious hypersensitivity reaction to dapagliflozin or hypersensitivity to metformin hydrochloride
- Acute or chronic metabolic acidosis, including diabetic ketoacidosis, with or without coma. Diabetic ketoacidosis should be treated with insulin
4.4 Special warnings and precautions for use
Lactic Acidosis
In Dapagliflozin + Metformin ER tablets a treated patient with a diagnosis or strong suspicion of lactic acidosis, prompt hemodialysis is recommended to correct the acidosis and remove accumulated metformin (metformin hydrochloride is dialyzable, with a clearance of up to 170 mL/min under good hemodynamic conditions). Hemodialysis has often resulted in reversal of symptoms and recovery. The postmarketing metformin-associated lactic acidosis cases primarily occurred in patients with significant renal impairment. The risk of metformin accumulation and metformin-associated lactic acidosis increases with the severity of renal impairment because metformin is substantially excreted by the kidney.
Hypotension
Dapagliflozin 2 causes intravascular volume contraction. Symptomatic hypotension can occur after initiating dapagliflozin, particularly in patients with impaired renal function (eGFR less than 60 mL/min/1.73 m ), elderly patients, or patients on loop diuretics. Before initiating Dapagliflozin + Metformin ER tablets in patients with one or more of these characteristics, volume status should be assessed and corrected. Monitor for signs and symptoms of hypotension after initiating therapy.
Ketoacidosis
Reports of ketoacidosis, a serious life-threatening condition requiring urgent hospitalization have been identified in postmarketing surveillance in patients with type 1 and type 2 diabetes mellitus taking sodium-glucose co transporter 2 (SGLT2) inhibitors, including dapagliflozin. Fatal cases of ketoacidosis have been reported in patients taking dapagliflozin. Dapagliflozin + Metformin ER tablet is not indicated for the treatment of patients with type 1diabetes mellitus
Acute Kidney Injury and Impairment in Renal Function
Dapagliflozin causes intravascular volume contraction and can cause renal impairment. There have been postmarketing reports of acute kidney injury, some requiring hospitalization and dialysis, in patients receiving dapagliflozin : some reports involved patients younger than 65 years of age. Before initiating Dapagliflozin + Metformin ER tablet, consider factors that may predispose patients to acute kidney injury including hypovolemia, chronic renal insufficiency, congestive heart failure, and concomitant medications (diuretics, ACE inhibitors, ARBs, NSAIDs). Consider temporarily discontinuing Dapagliflozin + Metformin ER tablet in any setting of reduced oral intake (such as acute illness or fasting) or fluid losses (gastrointestinal illness or excessive heat exposure); monitor patients for signs and symptoms of acute kidney injury. If acute kidney injury occurs, discontinue Dapagliflozin + Metformin ER tablet promptly and institute treatment.
4.5 Drugs interactions
Coadministration of multiple doses of dapagliflozin and metformin does not meaningfully alter the pharmacokinetics of either dapagliflozin or metformin in healthy subjects
No interaction studies have been performed for Dapaformin. The following statements reflect the information available on the individual active substances.
Dapagliflozin
Pharmacodynamic interactions
Diuretics
This medicinal product may add to the diuretic effect of thiazide and loop diuretics and may increase the risk of dehydration and hypotension.
Insulin and insulin secretagogues
Insulin and insulin secretagogues, such as sulphonylureas, cause hypoglycaemia. Therefore, a lower dose of insulin or an insulin secretagogue may be required to reduce the risk of hypoglycaemia when used in combination with dapagliflozin.
Pharmacokinetic interactions
The metabolism of dapagliflozin is primarily via glucuronide conjugation mediated by UDP-glucuronosyltransferase 1A9 (UGT1A9)
In vitro studies, dapagliflozin neither inhibited cytochrome P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, nor induced CYP1A2, CYP2B6 or CYP3A4. Therefore, this medicinal product is not expected to alter the metabolic clearance of coadministered medicinal products that are metabolised by these enzymes.
Effect of other medicinal products on dapagliflozin Interaction studies conducted in healthy subjects, using mainly a single-dose design, suggest that the pharmacokinetics of dapagliflozin are not altered by pioglitazone, sitagliptin, glimepiride, voglibose, hydrochlorothiazide, bumetanide, valsartan, or simvastatin.
Following coadministration of dapagliflozin with rifampicin (an inducer of various active transporters and drug-metabolising enzymes) a 22% decrease in dapagliflozin systemic exposure (AUC) was observed, but with no clinically meaningful effect on 24-hour urinary glucose excretion. No dose adjustment is recommended. A clinically relevant effect with other inducers (e.g. carbamazepine, phenytoin, phenobarbital) is not expected. Following coadministration of dapagliflozin with mefenamic acid (an inhibitor of UGT1A9), a 55% increase in dapagliflozin systemic exposure was seen, but with no clinically meaningful effect on 24-hour urinary glucose excretion. No dose adjustment is recommended.
Effect of dapagliflozin on other medicinal products In interaction studies conducted in healthy subjects, using mainly a single-dose design, dapagliflozin did not alter the pharmacokinetics of pioglitazone, sitagliptin, glimepiride, hydrochlorothiazide, bumetanide, valsartan, digoxin (a P-gp substrate) or warfarin (S-warfarin, a CYP2C9 substrate), or the anti-coagulatory effects of warfarin as measured by INR. Combination of a single dose of dapagliflozin 20 mg and simvastatin (a CYP3A4 substrate) resulted in a 19% increase in AUC of simvastatin and 31% increase in AUC of simvastatin acid. The increase in simvastatin and simvastatin acid exposures are not considered clinically relevant.
Interference with 1,5-anhydroglucitol (1,5-AG) assay Monitoring glycaemic control with 1,5-AG assay is not recommended as measurements of 1,5-AG are unreliable in assessing glycaemic control in patients taking SGLT2 inhibitors. Use of alternative methods to monitor glycaemic control is advised.
Metformin
Concomitant use not recommended Cationic substances that are eliminated by renal tubular secretion (e.g. cimetidine) may interact with metformin by competing for common renal tubular transport systems. A study conducted in seven normal healthy volunteers showed that cimetidine, administered as 400 mg twice daily, increased metformin systemic exposure (AUC) by 50% and Cmax by 81%. Therefore, close monitoring of glycaemic control, dose adjustment within the recommended posology and changes in diabetic treatment should be considered when cationic medicinal products that are eliminated by renal tubular secretion are coadministered.
Alcohol
Alcohol intoxication is associated with an increased risk of lactic acidosis, particularly in the case of fasting, malnutrition or hepatic impairment due to the metformin active substance of this medicinal product. Consumption of alcohol and medicinal products containing alcohol should be avoided.
Iodinated contrast agents
Intravascular administration of iodinated contrast agents may lead to contrast induced nephropathy, resulting in metformin accumulation and increased risk of lactic acidosis. Xigduo must be discontinued prior to, or at the time of the imaging procedure and not restarted until at least 48 hours after, provided that renal function has been re-evaluated and found to be stable.
Combination requiring precautions for use
Glucocorticoids (given by systemic and local routes), beta-2 agonists, and diuretics have intrinsic hyperglycaemic activity. The patient should be informed and more frequent blood glucose monitoring perfomed, especially at the beginning of treatment with such medicinal products. If necessary, the dose of the glucose-lowering medicinal product should be adjusted during therapy with the other medicinal product and on its discontinuation.
Some medicinal products can adversely affect renal function which may increase the risk of lactic acidosis, e.g. NSAIDs, including selective cyclo-oxygenase (COX) II inhibitors, ACE inhibitors, angiotensin II receptor antagonists and diuretics, especially loop diuretics. When starting or using such products in combination with metformin, close monitoring of renal function is necessary
Insulin and insulin secretagogues
Insulin and insulin secretagogues, such as sulphonylureas, cause hypoglycaemia. Therefore, a lower dose of insulin or an insulin secretagogue may be required to reduce the risk of hypoglycaemia when used in combination with metformin
4.6 Use in special populations
Pregnancy
Based on animal data showing adverse renal effects, Dapagliflozin + Metformin ER tablet is not recommended during the second and third trimesters of pregnancy. Limited data with Dapagliflozin + Metformin ER tablet or dapagliflozin in pregnant women are not sufficient to determine drug-associated risk for major birth defects or miscarriage. Published studies with metformin use during pregnancy have not reported a clear association with metformin and major birth defect or miscarriage risk. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy
Lactation
There is no information regarding the presence of Dapagliflozin + Metformin ER tablet or dapagliflozin in human milk, the effects on the breastfed infant, or the effects on milk production
Females and Males of Reproductive Potential Discuss the potential for unintended pregnancy with premenopausal women as therapy with metformin may result in ovulation in some anovulatory women.
Pediatric Use
Safety and effectiveness of Dapagliflozin + Metformin ER tablet in pediatric patients under 18 years of age have not been established.
Geriatric Use
No Dapagliflozin + Metformin ER tablet dosage change is recommended based on age. More frequent assessment of renal function is recommended in elderly patients.
Renal Impairment
In clinical studies dapagliflozin was associated with increases in serum creatinine and decreases in eGFR . Use of dapagliflozin is not recommended when eGFR is less than 45 mL/min/1.73 m and is contraindicated in patients with severe renal 2 impairment (eGFR less than 30 mL/min/1.73 m ) or ESRD.
Metformin is substantially excreted by the kidney, and the risk of metformin accumulation and lactic acidosis increases with the degree of renal impairment. Dapagliflozin + Metformin ER tablet is contraindicated in severe renal impairment, patients with 2 an estimated glomerular filtration rate (eGFR) below 30 mL/min/1.73 m
Hepatic Impairment
Use of metformin in patients with hepatic impairment has been associated with some cases of lactic acidosis. Dapagliflozin + Metformin ER tablet is not recommended in patients with hepatic impairment
4.7 Effects on ability to drive and use machines
Dapaformin has no or negligible influence on the ability to drive and use machines. Patients should be alerted to the risk of hypoglycaemia when this medicinal product is used in combination with other glucose-lowering medicinal products known to cause hypoglycaemia.
4.8 Undesirable effects
Dapaformin has been demonstrated to be bioequivalent with coadministered dapagliflozin and metformin. There have been no therapeutic clinical trials conducted with Dapaformin tablets.
Dapagliflozin plus metformin
Summary of the safety profile
In an analysis of 5 placebo-controlled dapagliflozin add-on to metformin studies, the safety results were similar to that of the pre-specified pooled analysis of 13 placebo-controlled dapagliflozin studies (see Dapagliflozin, Summary of the safety profile below). No additional adverse reactions were identified for the dapagliflozin plus metformin group compared with those reported for the individual components. In the separate dapagliflozin add-on to metformin pooled analysis, 623 subjects were treated with dapagliflozin 10 mg as add-on to metformin and 523 were treated with placebo plus metformin.
Dapagliflozin
Summary of the safety profile
In the clinical studies in type 2 diabetes, more than 15,000 patients have been treated with dapagliflozin.
The primary assessment of safety and tolerability was conducted in a pre-specified pooled analysis of 13 short-term (up to 24 weeks) placebo-controlled studies with 2,360 subjects treated with dapagliflozin 10 mg and 2,295 treated with placebo. In the dapagliflozin cardiovascular outcomes study (see section 5.1), 8,574 patients received dapagliflozin 10 mg and 8,569 received placebo for a median exposure time of 48 months. In total, there were 30,623 patient-years of exposure to dapagliflozin.
The most frequently reported adverse reactions across the clinical studies were genital infections.
Tabulated list of adverse reactions
The following adverse reactions have been identified in the placebo-controlled dapagliflozin plus metformin clinical studies, dapagliflozin clinical studies and metformin clinical studies and post-marketing experience. None were found to be doserelated. Adverse reactions listed below are classified according to frequency and system organ class. Frequency categories are defined according to the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), and not known (cannot be estimated from the available data).
Table : Adverse reactions in dapagliflozin and metformin immediate-release clinical trial and post-marketing data.
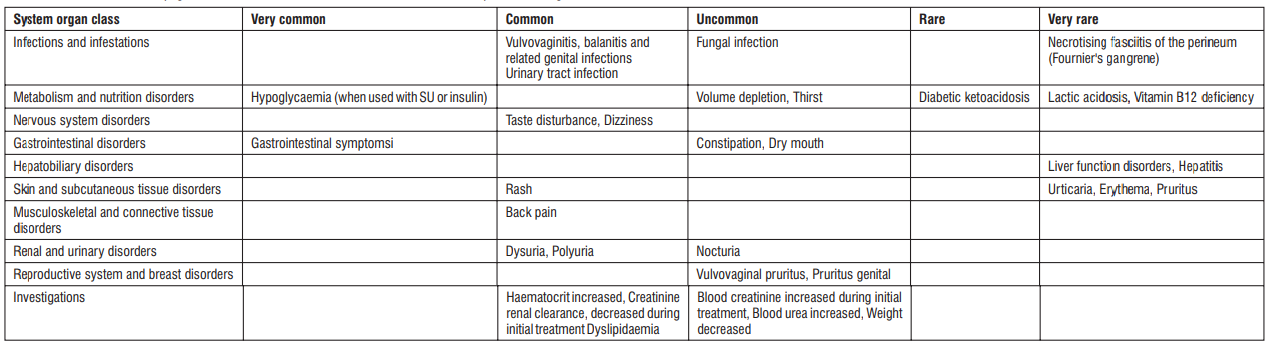
Description of selected adverse reactions
Dapagliflozin plus metformin
Hypoglycaemia
In studies with dapagliflozin in add-on combination with metformin, minor episodes of hypoglycaemia were reported at similar frequencies in the group treated with dapagliflozin 10 mg plus metformin (6.9%) and in the placebo plus metformin group (5.5%). No major events of hypoglycaemia were reported. Similar observations were made for the combination of dapagliflozin with metformin in drug-naive patients. In an add-on to metformin and a sulphonylurea study, up to 24 weeks, minor episodes of hypoglycaemia were reported in 12.8% of subjects who received dapagliflozin 10 mg plus metformin and a sulphonylurea and in 3.7% of subjects who received placebo plus metformin and a sulphonylurea. No major events of hypoglycaemia were reported.
Dapagliflozin
Vulvovaginitis, balanitis and related genital infections
n the 13-study safety pool, vulvovaginitis, balanitis and related genital infections were reported in 5.5% and 0.6% of subjects who received dapagliflozin 10 mg and placebo, respectively. Most infections were mild to moderate, and subjects responded to an initial course of standard treatment and rarely resulted in discontinuation from dapagliflozin treatment. These infections were more frequent in females (8.4% and 1.2% for dapagliflozin and placebo, respectively), and subjects with a prior history were more likely to have a recurrent infection.
In the dapagliflozin cardiovascular outcomes study, the number of patients with serious adverse events of genital infections were few and balanced : 2 patients in each of the dapagliflozin and placebo groups. Necrotising fasciitis of the perineum (Fournier's gangrene) Cases of Fournier's gangrene have been reported postmarketing in patients taking SGLT2 inhibitors, including dapagliflozin.
In the dapagliflozin cardiovascular outcomes study with 17,160 type 2 diabetes mellitus patients and a median exposure time of 48 months, a total of 6 cases of Fournier's gangrene were reported, one in the dapagliflozin-treated group and 5 in the placebo group.
Hypoglycaemia
The frequency of hypoglycaemia depended on the type of background therapy used in each study. For studies of dapagliflozin as add-on to metformin or as add-on to sitagliptin (with or without metformin), the frequency of minor episodes of hypoglycaemia was similar (< 5%) between treatment groups, including placebo up to 102 weeks of treatment. Across all studies, major events of hypoglycaemia were uncommon and comparable between the groups treated with dapagliflozin or placebo. In a study with add-on insulin therapy, higher rates of hypoglycaemia were observed. In an add-on to insulin study up to 104 weeks, episodes of major hypoglycaemia were reported in 0.5% and 1.0% of subjects in dapagliflozin 10 mg plus insulin at Weeks 24 and 104, respectively, and in 0.5% of subjects treated with placebo plus insulin groups at Weeks 24 and 104. At Weeks 24 and 104, minor episodes of hypoglycaemia were reported, respectively, in 40.3% and 53.1% of subjects who received dapagliflozin 10 mg plus insulin and in 34.0% and 41.6% of the subjects who received placebo plus insulin.
In the dapagliflozin cardiovascular outcomes study, no increased risk of major hypoglycaemia was observed with dapagliflozin therapy compared with placebo. Major events of hypoglycaemia were reported in 58 (0.7%) patients treated with dapagliflozin and 83 (1.0%) patients treated with placebo.
Volume depletion
In the 13-study safety pool, reactions suggestive of volume depletion (including, reports of dehydration, hypovolaemia or hypotension) were reported in 1.1% and 0.7% of subjects who received dapagliflozin 10 mg and placebo, respectively; serious reactions occurred in < 0.2% of subjects balanced between dapagliflozin 10 mg and placebo. In the dapagliflozin cardiovascular outcomes study, the numbers of patients with events suggestive of volume depletion were balanced between treatment groups: 213 (2.5%) and 207 (2.4%) in the dapagliflozin and placebo groups, respectively. Serious adverse events were reported in 81 (0.9%) and 70 (0.8%) in the dapagliflozin and placebo group, respectively. Events were generally balanced between treatment groups across subgroups of age, diuretic use, blood pressure and ACE-I/ARB 2 use. In patients with eGFR < 60 mL/min/1.73 m at baseline, there were 19 events of serious adverse events suggestive of volume depletion in the dapagliflozin group and 13 events in the placebo group. Diabetic ketoacidosis
In the dapagliflozin cardiovascular outcomes study, with a median exposure time of 48 months, events of DKA were reported in 27 patients in the dapagliflozin 10 mg group and 12 patients in the placebo group. The events occurred evenly distributed over the study period. Of the 27 patients with DKA events in the dapagliflozin group, 22 had concomitant insulin treatment at the time of the event. Precipitating factors for DKA were as expected in a type 2 diabetes mellitus population. Urinary tract infections
In the 13-study safety pool, urinary tract infections were more frequently reported for dapagliflozin compared with placebo (4.7% versus 3.5%, respectively). Most infections were mild to moderate, and subjects responded to an initial course of standard treatment and rarely resulted in discontinuation from dapagliflozin treatment. These infections were more frequent in females, and subjects with a prior history were more likely to have a recurrent infection. In the dapagliflozin cardiovascular outcomes study, serious events of urinary tract infections were reported less frequently for dapagliflozin 10 mg compared with placebo, 79 (0.9%) events versus 109 (1.3%) events, respectively. Increased creatinine
Adverse reactions related to increased creatinine were grouped (e.g. decreased renal creatinine clearance, renal impairment, increased blood creatinine and decreased glomerular filtration rate). This grouping of reactions was reported in 3.2% and 2 1.8% of patients who received dapagliflozin 10 mg and placebo, respectively. In patients with normal renal function or mild renal impairment (baseline eGFR ≥ 60 mL/min/1.73 m ) this grouping of reactions were reported in 1.3% and 0.8% of patients who 2 received dapagliflozin 10 mg and placebo, respectively. These reactions were more common in patients with baseline eGFR ≥ 30 and < 60 mL/min/1.73 m (18.5% dapagliflozin 10 mg vs 9.3% placebo). Further evaluation of patients who had renal-related adverse events showed that most had serum creatinine changes of ≤ 0.5 mg/dL from baseline. The increases in creatinine were generally transient during continuous treatment or reversible after discontinuation of treatment
In the dapagliflozin cardiovascular outcomes study, including elderly patients and patients with renal impairment (eGFR less than 60 mL/min/1.73 m ), eGFR decreased over time in both treatment groups. At 1 year, mean eGFR was slightly lower, and at 4 years, mean eGFR was slightly higher in the dapagliflozin group compared with the placebo group.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse
reactions via email to: medico@zuventus.com
By reporting side effects, you can help provide more information on the safety of this medicine
4.9 Overdose
Dapagliflozin
There were no reports of overdose during the clinical development program for dapagliflozin. In the event of an overdose, contact the Poison Control Center. It is also reasonable to employ supportive measures as dictated by the patient’s clinical status. The removal of dapagliflozin by hemodialysis has not been studied.
Metformin hydrochloride
Overdose of metformin hydrochloride has occurred, including ingestion of amounts >50 grams. Hypoglycemia was reported in approximately 10% of cases, but no causal association with metformin hydrochloride has been established. Lactic acidosis has been reported in approximately 32% of metformin overdose cases. Metformin is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions. Therefore, hemodialysis may be useful for removal of accumulated drug from patients in whom metformin overdosage is suspected.
5.0 Pharmacological properties
5.1 Mechanism of Action
Dapagliflozin + Metformin ER tablet combines two antihyperglycemic agents with complementary mechanisms of action to improve glycemic control in patients with type 2 diabetes: dapagliflozin, a sodiumglucose cotransporter 2 (SGLT2) inhibitor, and metformin hydrochloride, a biguanide. Dapagliflozin Sodium-glucose cotransporter 2 (SGLT2), expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Dapagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, and thereby increases urinary glucose excretion. Metformin hydrochloride Metformin improves glucose tolerance in patients with type 2 diabetes, lowering both basal and postprandial plasma glucose. Metfor min decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. Metformin does not produce hypoglycemia in either patients with type 2 diabetes or in healthy subjects, except in unusual circumstances, and does not cause hyperinsulinemia. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may actually decrease.
5.2 Pharmacodynamic properties
Dapagliflozin
Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in patients with type 2 diabetes mellitus following the administration of dapagliflozin Dapagliflozin doses of 5 or 10 mg per day in patients with type 2 diabetes mellitus for 12 weeks resulted in excretion of approximately 70 grams of glucose in the urine per day. A near maximum glucose excretion was observed at the dapagliflozin daily dose of 20 mg. This urinary glucose excretion with dapagliflozin also results in increases in urinary volume Dapagliflozin was not associated with clinically meaningful prolongation of QTc interval at daily doses up to 150 mg (15 times the recommended dose) in a study of healthy subjects. In addition, no clinically meaningful effect on QTc interval was observed following single doses of up to 500 mg (50 times the recommended dose) dapagliflozin in healthy subjects. Metformin In humans, independently of its action on glycaemia, metformin has favourable effects on lipid metabolism. This has been shown at therapeutic doses in controlled, medium-term or long-term clinical studies: metformin reduces total cholesterol, LDL cholesterol and triglyceride levels. In clinical studies, use of metformin was associated with either a stable body weight or modest weight loss.
5.3 Pharmacokinetic properties
Absorption
Dapagliflozin Following oral administration of dapagliflozin, the maximum plasma concentration (Cmax) is usually attained within 2 hours under fasting state. The Cmax and AUC values increase dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10 mg dose is 78%. Administration of dapagliflozin with a high-fat meal decreases its Cmax by up to 50% and prolongs Tmax by approximately 1 hour, but does not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful and dapagliflozin can be administered with or without food. Metformin Hydrochloride The absolute bioavailability of a 500 mg metformin hydrochloride tablet given under fasting conditions is approximately 50 to 60%. Studies using single oral doses of metformin hydrochloride tablets 500 mg to 1,500 mg, and 850 mg to 2,550 mg, indicate that there is a lack of dose proportionality with increasing doses, which is due to decreased absorption rather than an alteration in elimination. Food decreases the extent of and slightly delays the absorption of metformin hydrochloride, as shown by approximately a 40% lower mean peak plasma concentration (Cmax), a 25% lower area under the plasma concentration versus time curve (AUC), and a 35 minute prolongation of the time to peak plasma concentration (Tmax) following administration of a single 850 mg tablet of metformin hydrochloride with food, compared to the same tablet strength administered under fasting conditions. The clinical relevance of these decreases is unknown.
Distribution
Dapagliflozin Dapagliflozin is approximately 91% protein bound. Protein binding is not altered in patients with renal or hepatic impairment. Metformin Hydrochloride The apparent volume of distribution (V/F) of metformin hydrochloride following single oral doses of 850 mg averaged 654 ± 358 litres. Metformin hydrochloride is negligibly bound to plasma proteins, in contrast to sulphonylureas, which are more than 90% protein bound. Metformin hydrochloride partitions into erythrocytes, most likely as a function of time. At usual clinical doses and dosing schedules of metformin hydrochloride, steady state plasma concentrations of metformin hydrochloride are reached within 24 to 48 hours and are generally<1 microgram/mL. During controlled clinical studies of metformin hydrochloride, maximum metformin hydrochloride plasma levels did not exceed 5 micrograms/mL, even at maximum doses
Biotransformation
Dapagliflozin The metabolism of dapagliflozin is primarily mediated by UGT1A9; CYP-mediated metabolism is a minor clearance pathway in humans. Dapagliflozin is extensively metabolized, primarily to yield dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O glucuronide accounted for 61% of a 50 mg [14C]-dapagliflozin dose and is the predominant drugrelated component in human plasma. Metformin Hydrochloride Metformin is excreted unchanged in the urine. No metabolites have been identified in humans.
Elimination
Dapagliflozin and related metabolites are primarily eliminated via the renal pathway. Following a single 50 mg dose of [14C]-dapagliflozin, 75% and 21% total radioactivity is excreted in urine and feces, respectively. In urine, less than 2% of the dose is excreted as parent drug. In feces, approximately 15% of the dose is excreted as parent drug. The mean plasma terminal half-life (t½) for dapagliflozin is approximately 12.9 hours following a single oral dose of dapagliflozin 10 mg.
Metformin Hydrochloride
Intravenous single-dose studies in normal subjects demonstrate that metformin hydrochloride is excreted unchanged in the urine and does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion. Renal clearance is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of elimination. Following oral administration, approximately 90% of the absorbed drug is eliminated via the renal route within the first 24 hours, with a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
6.0 Nonclinical properties
No known animal toxicology data
7.0 Description
Dapagliflozin, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes. It is also used to treat adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.
The most common side effect in people with type 2 diabetes is hypoglycaemia, especially when used in combination with a sulphonylurea or insulin. The most common side effect in people with type 1 diabetes is genital infection, especially in women and a common side effect is diabetic ketoacidosis. It is of the gliflozin (SGLT2 inhibitor) class. Metformin, is the first-line medication for the treatment of type 2 diabetes, particularly in people who are overweight. It is also used in the treatment of polycystic ovary syndrome. It is not associated with weight gain. It is taken by mouth.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
No incompatibility study have been found
8.2 Shelf-life
Refer on the pack
8.3 Packaging information
A blister strip of 10 tablets.
8.4 Storage and handing instructions
Store below 30°C. Protect from light.
Keep out of reach of children
9.0 Patient counselling information
Instruct patients to read the Medication Guide before starting treatment with and to reread it each time the prescription is renewed. Inform patients of the potential risks and benefits of Dapaformin and of alternative modes of therapy. Also inform patients about the importance of adherence to dietary instructions, regular physical activity, periodic blood glucose monitoring and HbA1c testing, recognition and management of hypoglycemia and hyperglycemia, and assessment of diabetes complications. Advise patients to seek medical advice promptly during periods of stress such as fever, trauma, infection, or surgery, as medication requirements may change. Inform patients that the incidence of hypoglycemia may be increased when Dapaformin is added to an insulin secretagogue (e.g., sulfonylurea) or insulin. Instruct patient to immediately inform her healthcare provider if she is pregnant or plans to become pregnant. Based on animal data, Dapaformin may cause fetal harm in the second and third trimesters of pregnancy. Instruct patient to immediately inform her healthcare provider if she is breastfeeding or planning to breastfeed. It is not known if Dapaformin is excreted in breast milk; however, based on animal data, Dapaformin may cause harm to nursing infants. Inform patients that the most common adverse reactions associated with use of Dapaformin are female genital mycotic infections, nasopharyngitis, urinary tract infections, diarrhea, headache, nausea, and vomiting. Instruct patients that Dapaformin must be swallowed whole and not crushed or chewed, and that the inactive ingredients may occasionally be eliminated in the feces as a soft mass that may resemble the original tablet. Instruct patients to take Dapaformin only as prescribed. If a dose is missed, advise patients to take it as soon as it is remembered unless it is almost time for the next dose, in which case patients should skip the missed dose and take the medicine at the next regularly scheduled time. Advise patients not to take 2 tablets of Dapaformin at the same time, unless otherwise instructed by their healthcare provider.
12.0 Date of revision
02 June 2022

About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What DapaFormin 500/1000 is and what it is used for
- What you need to know before you take DapaFormin 500/1000
- How to take DapaFormin 500/1000 Possible side effects
- How to store DapaFormin 500/1000
- Contents of the pack and other information
1. What DapaFormin 500/1000 is and what it is used for
This medicine contains two different substances called dapagliflozin and metformin. Both belong to a group of medicines called oral anti-diabetics. These are medicines taken by mouth for diabetes.
DapaFormin 500/1000 is used for a type of diabetes called “type 2 diabetes” in adult patients (aged 18 years and older) and usually occurs when you are older. If you have type 2 diabetes, your pancreas does not make enough insulin or your body is not able to use the insulin it produces properly. This leads to a high level of sugar (glucose) in your blood.
- Dapagliflozin works by removing excess sugar from your body via your urine and lowers the amount of sugar in your blood. It can also help prevent heart disease.
- Metformin works mainly by inhibiting glucose production in the liver.
To treat diabetes:
- This medicine is taken in combination with diet and exercise.
- This medicine is used if your diabetes cannot be controlled with other medicines used to treat diabetes.
- Your doctor may ask you to take this medicine on its own or together with other medicines to treat diabetes.
- This may be another medicine taken by mouth and/or a medicine given by injection, such as insulin or a GLP-1 receptor agonist (helps your body to increase the production of insulin when your blood sugar is high).
- If you are already taking both dapagliflozin and metformin as single tablets, your doctor may ask you to switch to this medicine.
- To avoid overdose, do not continue taking dapagliflozin and metformin tablets, if you are taking DapaFormin 500/1000.
It is important to continue to follow the advice on diet and exercise given to you by your doctor, pharmacist or nurse.
2. What you need to know before you take DapaFormin 500/1000
Do not take DapaFormin 500/1000
- if you are allergic to dapagliflozin, metformin or any of the other ingredients of this medicine (listed in section 6).
- if you have ever had a diabetic coma.
- if you have uncontrolled diabetes, with, for example severe hyperglycaemia (high blood glucose), nausea, vomiting, diarrhoea, rapid weight loss, lactic acidosis (see “Risk of lactic acidosis” below) or ketoacidosis. Ketoacidosis is a condition in which substances called ‘ketone bodies’ accumulate in the blood and which can lead to a diabetic pre-coma. Symptoms include stomach pain, fast and deep breathing, sleepiness or your breath developing an unusual fruity smell.
- if you have severely reduced kidney function.
- if you have a severe infection.
- if you have lost a lot of water from your body (dehydration), e.g. due to long-lasting or severe diarrhoea, or if you have vomited several times in a row.
- if you have recently had a heart attack or if you have heart failure or serious problems with your blood circulation or difficulties in breathing.
- if you have problems with your liver.
- if you drink large amounts of alcohol, either every day or only from time to time (please see section “DapaFormin 500/1000 with alcohol”).
Do not take this medicine if any of the above apply to you.
Warnings and precautions
Risk of lactic acidosis
DapaFormin 500/1000 may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration (see further information below), liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease). If any of the above apply to you, talk to your doctor for further instructions.
Stop taking DapaFormin 500/1000 for a short time if you have a condition that may be associated with dehydration (significant loss of body fluids) such as severe vomiting, diarrhoea, fever, exposure to heat or if you drink less fluid than normal. Talk to your doctor for further instructions.
Stop taking DapaFormin 500/1000 and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma. Symptoms of lactic acidosis include:
- vomiting
- stomach ache (abdominal pain)
- muscle cramps
- a general feeling of not being well with severe tiredness - difficulty in breathing
- reduced body temperature and heartbeat
- Lactic acidosis is a medical emergency and must be treated in a hospital.
Talk to your doctor, pharmacist or nurse before taking DapaFormin 500/1000, and during treatment:
if you have “type 1 diabetes” – the type that usually starts when you are young, and your body does not produce any insulin. DapaFormin 500/1000 should not be used to treat this condition.
if you experience rapid weight loss, feeling sick or being sick, stomach pain, excessive thirst, fast and deep breathing, confusion, unusual sleepiness or tiredness, a sweet smell to your breath, a sweet or metallic taste in your mouth, or a different odour to your urine or sweat, contact a doctor or the nearest hospital straight away. These symptoms could be a sign of “diabetic ketoacidosis” – a rare but serious, sometimes life-threatening problem you can get with diabetes because of increased levels of “ketone bodies” in your urine or blood, seen in tests. The risk of developing diabetic ketoacidosis may be increased with prolonged fasting, excessive alcohol consumption, dehydration, sudden reductions in insulin dose, or a higher need of insulin due to major surgery or serious illness.
if you have problems with your kidneys. Your doctor will check your kidney function.
if you have very high levels of glucose in your blood which may make you dehydrated (lose too much body fluid). Possible signs of dehydration are listed at the top of section 4. Tell your doctor before you start taking this medicine if you have any of these signs.
if you are taking medicines to lower blood pressure (anti-hypertensives) and have a history of low blood pressure (hypotension). More information is given below under ‘Other medicines and DapaFormin 500/1000’.
if you often get infections of the urinary tract. This medicine may cause urinary tract infections and your doctor may want to monitor you more closely. Your doctor may consider temporarily changing your treatment if you develop a serious infection.
If you need to have major surgery, you must stop taking DapaFormin 500/1000 during and for some time after the procedure. Your doctor will decide when you must stop and when to restart your treatment with DapaFormin 500/1000.
It is important to check your feet regularly and adhere to any other advice regarding foot care given by your health care professional.
If any of the above applies to you (or you are not sure), talk to your doctor, pharmacist or nurse before taking this medicine.
Talk to your doctor immediately if you develop a combination of symptoms of pain, tenderness, redness, or swelling of the genitals or the area between the genitals and the anus with fever or feeling generally unwell. These symptoms could be a sign of a rare but serious or even life-threatening infection, called necrotising fasciitis of the perineum or Fournier’s gangrene which destroys the tissue under the skin. Fournier’s gangrene has to be treated immediately.
Kidney function
During treatment with DapaFormin 500/1000, your doctor will check your kidney function at least once every year or more frequently if you are elderly and/or if you have worsening kidney function.
Urine glucose
Because of how this medicine works, your urine will test positive for sugar while you are on this medicine.
Children and adolescents
This medicine is not recommended for children and adolescents under 18 years of age, because it has not been studied in these patients.
Other medicines and DapaFormin 500/1000
If you need to have an injection of a contrast medium that contains iodine into your bloodstream, for example in the context of an X-ray or scan, you must stop taking DapaFormin 500/1000 before or at the time of the injection. Your doctor will decide when you must stop and when to restart your treatment with DapaFormin 500/1000.
Tell your doctor if you are taking, have recently taken or might take any other medicines. You may need more frequent blood glucose and kidney function tests, or your doctor may adjust the dosage of DapaFormin 500/1000. It is especially important to mention the following:
- if you are taking medicines which increase urine production (diuretics).
- Your doctor may ask you to stop taking this medicine. Possible signs of losing too much fluid from your body are listed at the top of section 4.
- if you are taking other medicines that lower the amount of sugar in your blood such as insulin or a “sulphonylurea” medicine.
- Your doctor may want to lower the dose of these other medicines, to prevent you from getting blood sugar levels that are too low (hypoglycaemia). if you are taking cimetidine, a medicine used to treat stomach problems.
- if you are using bronchodilators (beta-2 agonists) which are used to treat asthma.
- if you are using corticosteroids (used to treat inflammation in diseases like asthma and arthritis) that are given by mouth, as an injection, or inhaled.
- if you are using medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as ibuprofen and celecoxib).
- if you are using certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists).
DapaFormin 500/1000 with alcohol
Avoid excessive alcohol intake while taking DapaFormin 500/1000 since this may increase the risk of lactic acidosis (see “Warnings and precautions”).
Pregnancy and breast-feeding
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine. You should stop taking this medicine if you become pregnant, since it is not recommended during the second and third trimesters (the last six months) of pregnancy. Talk to your doctor about the best way to control your blood sugar while you are pregnant.
Talk to your doctor if you would like to or are breast-feeding before taking this medicine. You should not use this medicine if you are breast-feeding. Metformin passes into human milk in small amounts. It is not known if dapagliflozin passes into human breast milk.
Driving and using machines
This medicine has no or negligible influence on the ability to drive and use machines. Taking it with other medicines that lower the amount of sugar in your blood, such as insulin or a “sulphonylurea” medicine, can cause too low blood sugar levels (hypoglycaemia), which may cause symptoms such as weakness, dizziness, increased sweating, fast heart beat, change in vision or difficulties concentrating, and may affect your ability to drive and use machines. Do not drive or use any tools or machines, if you start to feel these symptoms.
Sodium content
This medicine contains less than 1 mmol sodium (23 mg) per dose, that is to say it is essentially ‘sodiumfree’.
3. How to take DapaFormin 500/1000
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
How much to take
- The amount of this medicine that you will take varies depending on your condition and the doses you currently take of metformin and/or individual tablets of dapagliflozin and metformin. Your doctor will tell you exactly which strength of this medicine to take.
- The recommended dose is one tablet twice a day.
Taking this medicine
- Swallow the tablet whole with half a glass of water.
- Take your tablet with food. This is to reduce the risk of side effects in the stomach.
- Take your tablet twice daily, once in the morning (breakfast) and once in the evening (dinner).
Your doctor may prescribe this medicine together with other medicine(s) to lower the amount of sugar in your blood. These may be medicine(s) by mouth or given by injection, such as insulin or a GLP-1 receptor agonist. Remember to take these other medicine(s) as your doctor has told you. This will help get the best results for your health.
Diet and exercise
To control your diabetes, you still need to keep to diet and exercise, even when you are taking this medicine. So it is important to keep following the advice about diet and exercise from your doctor, pharmacist or nurse. In particular, if you are following a diabetic weight control diet, continue to follow it while you are taking this medicine.
If you take more DapaFormin 500/1000 than you should
If you take more DapaFormin 500/1000 tablets than you should, you may experience lactic acidosis. Symptoms of lactic acidosis include feeling or being very sick, vomiting, stomach ache, muscular cramps, severe tiredness or difficulty breathing. If this happens to you, you may need immediate hospital treatment, as lactic acidosis may lead to coma. Stop taking this medicine immediately and contact a doctor or the nearest hospital straight away (see section 2). Take the medicine pack with you.
If you forget to take DapaFormin 500/1000
If you miss a dose, take it as soon as you remember. If you do not remember until it is time for your next dose, skip the missed dose and go back to your regular schedule. Do not take a double dose of this medicine to make up for a forgotten dose.
If you stop taking DapaFormin 500/1000
Do not stop taking this medicine without talking to your doctor first. Your blood sugar may increase without this medicine.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Stop taking DapaFormin 500/1000 and see a doctor straight away if you notice any of the following serious or potentially serious side effects:
Lactic acidosis, seen very rarely (may affect up to 1 in 10,000 people) DapaFormin 500/1000 may cause a very rare, but very serious side effect called lactic acidosis (see section “Warnings and precautions”). If this happens you must stop taking DapaFormin
500/1000 and contact a doctor or the nearest hospital immediately, as lactic acidosis may lead to coma
Contact a doctor or the nearest hospital straight away if you have any of the following side effects:
Diabetic ketoacidosis, seen rarely (may affect up to 1 in 1,000 people) These are the signs of diabetic ketoacidosis (see also section 2 Warnings and precautions): - increased levels of “ketone bodies” in your urine or blood
rapid weight loss feeling sick or being sick stomach pain excessive thirst fast and deep breathing confusion unusual sleepiness or tiredness a sweet smell to your breath, a sweet or metallic taste in your mouth or a different odour to your urine or sweat.
This may occur regardless of blood glucose level. Your doctor may decide to temporarily or permanently stop your treatment with DapaFormin 500/1000.
Stop taking DapaFormin 500/1000 and see a doctor as soon as possible if you notice any of the following serious or potentially serious effects:
Dehydration: loss of too much fluid from your body, seen uncommonly (may affect up to 1 in 100 people).
These are signs of dehydration:
very dry or sticky mouth, feeling very thirsty feeling very sleepy or tired - passing little or no water (urine) - fast heartbeat.
Urinary tract infection, seen commonly (may affect up to 1 in 10 people). These are signs of a severe infection of the urinary tract: - fever and/or chills
burning sensation when passing water (urinating) - pain in your back or side. Although uncommon, if you see blood in your urine, tell your doctor immediately.
Contact your doctor as soon as possible if you have any of the following side effects:
Low blood sugar levels (hypoglycaemia), seen very commonly (may affect more than 1 in 10 people) - when taking this medicine with a sulphonylurea or other medicines that lower the amount of sugar in your blood, such as insulin.
These are the signs of low blood sugar:
- shaking, sweating, feeling very anxious, fast heart beat - feeling hungry, headache, change in vision
- a change in your mood or feeling confused.
Your doctor will tell you how to treat low blood sugar levels and what to do if you get any of the signs above. If you have symptoms of low blood sugar, eat glucose tablets, a high sugar snack or drink fruit juice. Measure your blood sugar if possible and rest.
Other side effects include:
Very common
- nausea, vomiting
- diarrhoea or stomach ache
- loss of appetite
Common
- genital infection (thrush) of your penis or vagina (signs may include irritation, itching, unusual discharge or odour)
- back pain
- discomfort when passing water (urine), passing more water than usual or needing to pass water more often
- changes in the amount of cholesterol or fats in your blood (shown in tests)
- increases in the amount of red blood cells in your blood (shown in tests)
- decreases in creatinine renal clearance (shown in tests) in the beginning of treatment
- changes in taste dizziness rash
Uncommon
- thirst
- constipation
- awakening from sleep at night to pass urine
- dry mouth
- weight decreased
- increases in creatinine (shown in laboratory blood tests) in the beginning of treatment
- increases in urea (shown in laboratory blood tests)
Very rare
- decreased vitamin B12 levels in the blood
- abnormalities in liver function tests, inflammation of the liver (hepatitis) • redness of the skin (erythema), itching or an itchy rash (hives)
Reporting of side effects
If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top right end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
5. How to store DapaFormin 500/1000
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the blister or carton after ‘EXP’. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What DapaFormin 500/1000 contains
DapaFormin 500
Each film coated tablet contains :
Monohydrate
equivalent to Dapagliflozin 10 mg
Excipients q.s.
Colours : Titanium Dioxide IP & Ferric Oxide Yellow USP-NF
DapaFormin 1000
Each film coated tablet contains :
Dapagliflozin Propanediol
equivalent to Dapagliflozin 10 mg
Metformin Hydrochloride IP 1000 mg
(Extended Release)
Excipients q.s.
For More Information About This Product
Zuvog M 0.3 mg Tablet
1.0 Generic Name
Voglibose and Metformin Hydrochloride (Prolonged-release) Tablets
2.0 Qualitative and quantitative composition
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
3.0 Dosage form and strength
Dosage Form: Tablets.
Dosage Strength: Voglibose 0.2 mg/ 0.3 mg and Metformin Hydrochloride 500 mg
(Prolonged-release)
4.0 Clinical particulars
4.1 Therapeutic Indication
As 2nd line treatment of type II Diabetes mellitus when diet, exercise and voglibose or metformin do not result in adequate glycemic control individually.
4.2 Posology and method of administration
Dosage should be individualised on the basis of both effectiveness and tolerance.
The initial recommended dose is one tablet of ZUVOG M 0.2 three times daily just before each meal.
In case of inadequate effect, the dose may be increased up to 1 tablet of ZUVOG M 0.3, three times daily just before each meal under close observation of the course of the disease.
4.3 Contraindications
- Renal disease or renal dysfunction (e.g., as suggested by serum creatinine levels 1.5 mg/dL [males], 1.4 mg/dL [females] or abnormal creatinine clearance), which may also result from conditions such as cardiovascular collapse (shock), acute myocardial infarction, and septicaemia.
- Hypersensitivity to metformin, voglibose or any of the components of this product.
- Acute or chronic metabolic acidosis, including diabetic ketoacidosis, with or without coma. Diabetic ketoacidosis should be treated with insulin.
- Severe infection, before and after surgery, serious trauma.
- Gastrointestinal obstruction or predisposed to it.
Patients undergoing radiologic studies involving intravascular administration of iodinated contrast materials should temporarily discontinue taking this combination, because use may result in acute alteration of renal function.
4.4 Special warnings and precautions for use
Loss of Control of Blood Glucose
When a patient stabilised on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of glycaemic control may occur. At such times, it may be necessary to withhold oral antidiabetic agents and temporarily administer insulin. This combinationmay be reinstituted after the acute episode is resolved.
Hypoxic States
Cardiovascular collapse (shock) from whatever cause, acute congestive heart failure, acute myocardial infarction and other conditions characterised by hypoxaemia have been associated with lactic acidosis and may also cause prerenalazotaemia. When such events occur in patients on ZUVOG M, the drug should be promptly discontinued.
Surgical Procedures
ZUVOG M should be temporarily suspended for any surgical procedure (except minor procedures not associated with restricted intake of food and fluids) and should not be restarted until the patient’s oral intake has resumed and renal function has been evaluated as normal.
Alcohol Intake
Alcohol is known to potentiate the effect of metformin on lactate metabolism. Patients, therefore, should be warned against excessive alcohol intake, acute or chronic, while receiving ZUVOG M.
Change in Clinical Status of Patients with Previously Controlled Type 2 Diabetes
A patient with type 2 diabetes with previously well controlled type 2 diabetes who develops laboratory abnormalities or clinical illness (especially vague and poorly defined illness) should be evaluated promptly for evidence of ketoacidosis or lactic acidosis. Evaluation should include serum electrolytes and ketones, blood glucose and, if indicated, blood pH, lactate, pyruvate, and metformin levels. If acidosis of either form occurs, ZUVOG M must be stopped immediately and other appropriate corrective measures initiated.
Hypoglycaemia
Hypoglycaemia does not occur in patients receiving ZUVOG M alone under usual circumstances of use, but could occur when caloric intake is deficient, when strenuous exercise is not compensated by caloric supplementation, or during concomitant use with other glucose-lowering agents (such as sulphonylureas and insulin) or ethanol. Elderly, debilitated, or malnourished patients and those with adrenal or pituitary insufficiency or alcohol intoxication are particularly susceptible to hypoglycaemic effects. Hypoglycaemia may be difficult to recognize in the elderly, and in people who are taking beta-adrenergic blocking drugs. Patients should be instructed and explained to recognize hypoglycaemic symptoms and its management.
Vitamin B12
In controlled clinical trials of metformin of 29 weeks duration, a decrease to subnormal levels of previously normal serum vitamin B12 levels, without clinical manifestations, was observed in approximately 7% of patients. Such decrease, possibly due to interference with B12 absorption from the B12 intrinsic factor complex, however is very rarely associated with anaemia and appears to be rapidly reversible with discontinuation of metformin or vitamin B12 supplementation. Measurement of haematologic parameters on an annual basis is advised in patients on metformin and any apparent abnormalities should be appropriately investigated and managed.
Certain individuals (those with inadequate vitamin B12 or calcium intake or absorption) appear to be predisposed to developing subnormal vitamin B12 levels. In these patients, routine serum vitamin B12 measurements at 2- or 3- year intervals may be useful.
Macrovascular Outcomes
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with voglibose, metformin or any other anti-diabetic drug.
Lactic Acidosis
Lactic acidosis is a rare, but serious, metabolic complication that can occur due to metformin accumulation during treatment with metformin; when it occurs, it is fatal in approximately 50% of cases. Lactic acidosis may also occur in association with a number of pathophysiologic conditions, including diabetes mellitus, and whenever there is significant tissue hypo perfusion and hypoxaemia. Lactic acidosis is characterised by elevated blood lactate levels (>5 mmol/L), decreased blood pH, electrolyte disturbances with an increased anion gap, and an increased lactate/pyruvate ratio. When metformin is implicated as the cause of lactic acidosis, metformin plasma levels >5 μg/mL are generally found.
The reported incidence of lactic acidosis in patients receiving metformin hydrochloride is very low. Patients with congestive heart failure requiring pharmacologic management, in particular those with unstable or acute congestive heart failure who are at risk of hypo perfusion and hypoxaemia, are at increased risk of lactic acidosis. The risk of lactic acidosis increases with the degree of renal dysfunction and the patient’s age. The risk of lactic acidosis may, therefore, be significantly decreased by regular monitoring of renal function in patients taking metformin and by use of the minimum effective dose of metformin. In particular, treatment of the elderly should be accompanied by careful monitoring of renal function. Metformin treatment should not be initiated in patients ≥80 years of age unless measurement of creatinine clearance demonstrates that renal function is not reduced, as these patients are more susceptible to developing lactic acidosis. In addition, metformin should be promptly withheld in the presence of any condition associated with hypoxaemia, dehydration, or sepsis. Because impaired hepatic function may significantly limit the ability to clear lactate, metformin should generally be avoided in patients with clinical or laboratory evidence of hepatic disease. Patients should be cautioned against excessive alcohol intake, either acute or chronic, when taking metformin, since alcohol potentiates the effects of metformin hydrochloride on lactate metabolism. In addition, metformin should be temporarily discontinued prior to any intravascular radiocontrast study and for any surgical procedure.
The onset of lactic acidosis often is subtle, and accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, increasing somnolence, and nonspecific abdominal distress. There may be associated hypothermia, hypotension, and resistant bradyarrhythmias with more marked acidosis. The patient and the patient’s physician must be aware of the possible importance of such symptoms and the patient should be instructed to notify the physician immediately if they occur. Metformin should be withdrawn until the situation is clarified. Serum electrolytes, ketones, blood glucose, and if indicated, blood pH, lactate levels, and even blood metformin levels may be useful. Once a patient is stabilised on any dose level of metformin, gastrointestinal symptoms, which are common during initiation of therapy, are unlikely to be drug related. Later occurrence of gastrointestinal symptoms could be due to lactic acidosis or other serious disease.
Levels of fasting venous plasma lactate above the upper limit of normal but less than 5 mmol/L in patients taking metformin do not necessarily indicate impending lactic acidosis and may be explainable by other mechanisms, such as poorly controlled diabetes or obesity, vigorous physical activity, or technical problems in sample handling.
Lactic acidosis should be suspected in any diabetes patient with metabolic acidosis lacking evidence of ketoacidosis (ketonuria and ketonemia).
Lactic acidosis is a medical emergency that must be treated in a hospital setting. In a patient with lactic acidosis who is taking metformin, the drug should be discontinued immediately and general supportive measures promptly instituted. Because metformin hydrochloride is dialyzable (with a clearance of up to 170 mL/min under good hemodynamic conditions), prompt haemodialysis is recommended to correct the acidosis and remove the accumulated metformin. Such management often results in prompt reversal of symptoms and recovery.
4.5 Drugs interactions
Cationic Drugs
Certain medications used concomitantly with metformin may increase the risk of lactic acidosis. Cationic drugs that are eliminated by renal tubular secretions (e.g: amiloride, cimetidine, digoxin, morphine, procainamide, quinidine, quinine, ranitidine, triamterene, trimethoprim or vancomycin) may decrease metformin elimination by competing for common renal tubular transport systems. Hence, careful patient monitoring and dose adjustment of ZUVOG M and/or interfering drug is recommended in patients who are taking cationic medications that are excreted via the proximal renal tubular secretory system.
Furosemide
A single-dose, metformin-furosemide drug interaction study in healthy subjects demonstrated that pharmacokinetic parameters of both compounds were affected by co-administration. Furosemide increased the metformin plasma and blood Cmax by 22% and blood AUC by 15%, without any significant change in metformin renal clearance. When administered with metformin, the Cmax and AUC of furosemide were 31% and 12% smaller, respectively, than when administered alone, and the terminal half-life was decreased by 32%, without any significant change in furosemide renal clearance. No information is available about the interaction of metformin and furosemide when co-administered chronically.
Nifedipine
Nifedipine appears to enhance the absorption of metformin, it increases plasma metformin Cmax and AUC by 20% and 9% respectively and increases the amount of metformin excreted in the urine. Metformin has minimal effects on nifedipine.
Danazol
If the use of this active substance cannot be avoided, warn the patients and emphasise the importance of urine and blood glucose monitoring. It may be necessary to adjust the dose of voglibose and metformin during and after treatment with danazol.
Salicylates
If salicylates are administered or discontinued in patients receiving oral antidiabetic agents, patients should be monitored for hypoglycaemia or loss of blood glucose control.
Thiazide
Interactions between thiazide diuretics and oral antidiabetic agent’s decreases insulin sensitivity thereby leading to glucose intolerance and hyperglycaemia, thus leading to a loss of diabetic control. Hence diabetic patients should be monitored closely.
Other
Concomitant administration of angiotensin enzyme inhibitors (captopril, enalapril), other antidiabetic drugs (insulin, acarbose) beta-blockers, fluconazole, histamine (H2) receptor antagonist, monoamine oxidase inhibitors (MAOIs), sulphonamides and non-steroidal anti-inflammatory agents increase sensitivity to insulin and potentiation of blood glucose lowering effect and thus, in some instances, hypoglycaemia may occur . Certain drugs tend to produce hyperglycaemia and may lead to loss of glycaemic control. These drugs include corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel blocking drugs, quinolones and isoniazid. Patients receiving these drugs should be closely monitored for loss of diabetic control when therapy is instituted or discontinued. Dosage of the oral antidiabetic agents may need to be reduced.
Drugs Enhancing (beta-blockers, salicylic acid preparations, MAOs, fibrate derivatives) or Diminishing (epinephrine, adrenocortical hormone, thyroid hormone etc.) the Hypoglycaemic Action of Anti-Diabetic Drugs
When ZUVOG M is administered concomitantly with drugs that enhance or diminish the hypoglycaemic action of anti-diabetic drugs, caution should be taken as this might additionally delay the action of voglibose on the absorption of carbohydrates.
Warfarin
Metformin and voglibose do not affect the pharmacokinetics of warfarin, hence ZUVOG M can be safely administered along with warfarin.
4.6 Use in special populations
Pregnant Women
Recent information suggests that abnormal blood glucose levels during pregnancy are associated with the higher incidence of congenital abnormalities. There are no adequate and well-controlled studies in pregnant women with metformin. Metformin was not teratogenic in rats and rabbits at doses up to 600 mg/kg/day. The safety and effectiveness of voglibose in pregnant women has not been established. Animal studies have shown that voglibose is transferred to the foetus. ZUVOG M should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus. Most experts suggest insulin be used to maintain the blood glucose levels as close to normal as possible.
Lactating Women
Studies in lactating rats show that metformin is excreted into milk and reaches levels comparable to those in plasma. Similar studies have not been conducted on nursing mothers. Animal studies have shown a suppressive action of voglibose on body weight increase in newborns, mainly due to suppression of milk production resulting from inhibition of carbohydrate absorption in mother animals. Nursing should be discontinued if ZUVOG M has to be administered. If the use of this combination is discontinued, and if the diet alone is inadequate for controlling blood glucose, insulin therapy should be considered.
Paediatric Patients
The safety and effectiveness of metformin for the treatment of type 2 diabetes have been established in paediatric patients ages 10 to 16 years (studies have not been conducted in paediatric patients below the age of 10 years). The safety and effectiveness of voglibose in children has not been established. Hence ZUVOG M should not be considered in this population.
Geriatric
Metformin is known to be excreted through the kidneys and because risk of serious adverse reactions to the drug is greater in patients with impaired renal function, ZUVOG M should be used only in patients with normal renal function. Because aging is associated with reduced renal function, the use of ZUVOG M should be with caution as age increases. Care should be taken in the dose selection and regular renal function be monitored. The administration of ZUVOG M should be initiated at a lower dose in elderly patients and generally, should not be titrated to the maximum dose. The combination should be carefully administered under close observation of the course of disease conditions, with careful attention to the blood sugar level and the onset of gastrointestinal symptoms.
Renal Impairment Patients
Metformin is known to be substantially excreted by the kidney and the risk of metformin accumulation and lactic acidosis increases with the degree of impairment of renal function. Thus, patients with serum creatinine levels above the upper limit of normal for their age should not receive ZUVOG M. In patients with advanced age, ZUVOG M should be carefully titrated to establish the minimum dose for adequate glycaemic effect, because aging is associated with reduced renal function. In elderly patients, particularly those ≥80 years of age, renal function should be monitored regularly and, generally, ZUVOG M should not be titrated to the maximum dose. Before initiation of ZUVOG M and at least annually thereafter, renal function should be assessed and verified as normal. In patients in whom development of renal dysfunction is anticipated, renal function should be assessed more frequently and ZUVOG M discontinued if evidence of renal impairment is present.
4.7 Effects on ability to drive and use machines
This combination containing voglibose and metformin does not cause hypoglycaemia and therefore has no effect on the ability to drive or to use machines. However, patients should be alerted to the risk of hypoglycaemia when ZUVOG M is used in combination with other antidiabetic agents (e.g. sulphonylureas, insulin, or meglinitides).
4.8 Undesirable effects
Voglibose
The gastrointestinal adverse effects like diarrhoea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting or heartburn may occur with the use of voglibose. Abdominal swelling, increased flatus, and intestinal obstruction like symptoms due to an increase in intestinal gas, etc. may occur. Serious hepatic dysfunction accompanied with jaundice, increased aspartate aminotransferase (AST) or alanine aminotransferase (ALT), etc. may also occur. One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported; a causal relationship appears likely. When voglibose is administered to the patients with serious liver cirrhosis, hyperammonia may worsen with the development of constipation, etc., followed by disturbance of consciousness. When voglibose is used in combination with other antidiabetic drugs, hypoglycaemia may occur.
Metformin
The most common adverse reactions of metformin hydrochloride are diarrhoea, nausea/vomiting, flatulence, asthenia, indigestion, abdominal discomfort, headache, abnormal stools, hypoglycaemia, myalgia, lightheaded, dyspnea, nail disorder, rash, sweating increased, taste disorder, chest discomfort, chills, flu syndrome, flushing and palpitation. The very rare adverse effects include isolated reports of liver function tests, abnormalities or hepatitis resolving upon metformin discontinuation and skin reactions such as erythema, pruritus, urticaria.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
4.9 Overdose
Overdose of metformin hydrochloride has occurred, including ingestion of amounts greater than 50 grams. Hypoglycaemia was reported in approximately 10% of cases, but no causal association with metformin hydrochloride has been established. Lactic acidosis has been reported in approximately 32% of metformin overdose cases. Metformin is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions. Therefore, haemodialysis may be useful for removal of accumulated drug from patients in whom metformin overdosage is suspected.
Voglibose competitively and reversibly inhibits the alpha-glucosidase enzymes in the brush border in the small intestine, which delays the hydrolysis of complex carbohydrates. It appears unlikely to produce hypoglycaemia in overdose, but abdominal discomfort and diarrhea may occur.
5.0 Pharmacological properties
5.1 Pharmacodynamic/ Mechanism of Action
Voglibose
Voglibose is an alpha glucosidase inhibitor which inhibits the activity of alpha glucosidases that catalyse the decomposition of disaccharides into monosaccharides in the intestine, thereby delaying the digestion and absorption of carbohydrates, resulting in improvement of postprandial hyperglycaemia.
Metformin Hydrochloride
Metformin improves glucose tolerance in patients with type 2 diabetes (NIDDM), lowering both basal and postprandial plasma glucose. Metformin is not chemically or pharmacologically related to sulphonylureas, thiazolidinediones, or alpha-glucosidase inhibitors. Metformin decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. Unlike sulphonylureas, metformin does not produce hypoglycaemia in either patient with type 2 diabetes or normal subjects and does not cause hyperinsulinaemia. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may actually decrease.
5.2 Pharmacokinetic properties
Voglibose
Voglibose is poorly absorbed after oral doses. Plasma concentrations after oral doses have usually been undetectable. Following repeated administration to healthy subjects (n=6) in a single dose of 0.2 mg, 3 times a day, for 7 consecutive days, voglibose was not detected in plasma or urine. Similarly, when voglibose was administered to healthy male adults (n=10) as a single dose of 2 mg, voglibose was not detected in plasma or urine. After ingestion of voglibose, the majority of active unchanged drug remains in the lumen of the gastrointestinal tract to exert its pharmacological activity. Voglibose is metabolized by intestinal enzymes and by the microbial flora. Voglibose is mainly excreted in the feces.
Metformin Hydrochloride
The absolute bioavailability of a metformin 500-mg tablet given under fasting conditions is approximately 50-60%. Studies using single oral doses of metformin 500 mg to 1500 mg, and 850 mg to 2550 mg, indicate that there is a lack of dose proportionality with increasing doses, which is due to decreased absorption rather than an alteration in elimination. Food decreases the extent of and slightly delays the absorption of metformin, as shown by approximately a 40% lower mean peak plasma concentration (Cmax), a 25% lower area under the plasma concentration versus time curve (AUC), and a 35-minute prolongation of time to peak plasma concentration (Tmax) following administration of a single 850-mg tablet of metformin with food, compared to the same tablet strength administered fasting. The clinical relevance of these decreases is unknown.
The apparent volume of distribution (V/F) of metformin following single oral doses of metformin immediate-release 850 mg averaged 654 ± 358 L. Metformin is negligibly bound to plasma proteins, in contrast to sulphonylureas, which are more than 90% protein bound. Metformin partitions into erythrocytes, most likely as a function of time. At usual clinical doses and dosing schedules of metforminimmediate-release, steady state plasma concentrations of metformin are reached within 24-48 hours and are generally <1 µg/mL. During controlled clinical trials of immediate-release metformin, maximum metformin plasma levels did not exceed 5 µg/mL, even at maximum doses.
Intravenous single-dose studies in normal subjects demonstrate that metformin is excreted unchanged in the urine and does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion. Renal clearance of metformin is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of metformin elimination. Following oral administration, approximately 90% of the absorbed drug is eliminated via the renal route within the first 24 hours, with a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Voglibose
A study demonstrated that voglibose can potentiate CCl4 (carbon tetrachloride) and APAP (acetaminophen) hepatotoxicity in rats by inducing hepatic CYP2E1.
Metformin
Long-term carcinogenicity studies have been performed in rats (dosing duration of 104 weeks) and mice (dosing duration of 91 weeks) at doses up to and including 900 mg/kg/day and 1500 mg/kg/day, respectively. These doses are both approximately 3 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons. No evidence of carcinogenicity with metformin was found in either male or female mice. Similarly, there was no tumorigenic potential observed with metformin in male rats. There was, however, an increased incidence of benign stromal uterine polyps in female rats treated with 900 mg/kg/day.
There was no evidence of a mutagenic potential of metformin in the following in vitro tests: Ames test (S. typhimurium), gene mutation test (mouse lymphoma cells), or chromosomal aberrations test (human lymphocytes). Results in the in vivo mouse micronucleus test were also negative.
Fertility of male or female rats was unaffected by metformin when administered at doses as high as 600 mg/kg/day, which is approximately 2 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons.
7.0 Description
Zuvog M contains two oral antihyperglycemic drugs, voglibose and metformin hydrochloride, used in the management of type 2 diabetes mellitus.

8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
Zuvog M 0.2 / 0.3 - A blister strip of 10 tablets
8.4 Storage and handing instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient Counselling Information
- Tablet should be swallowed whole & not to be broken, crushed or chewed.
- Take this medicine exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting your doctor.
- Tell your doctor immediately if you experience any deep or rapid breathing, persistent nausea, vomiting, and stomach pain as ZUVOG M Tablet may cause a rare but serious condition called lactic acidosis, which is an excess of lactic acid in the blood.
- Pregnant women and breastfeeding mothers should not use this medicine without doctor consultation.
- Don’t medicine for the treatment of diabetic ketoacidosis or diabetic pre-coma.
- This medicine is not advisable for use in children.
- Avoid use of this medicine during severe infection or before and after surgery or in serious trauma cases.
- This tablet may cause hypoglycemia when co-administered with oral sulfonylureas and insulin.
12.0 Date of revision
11/10/2024
The name of your medicine is ZUVOG M. We refer to them as ZUVOG M TABLETS or ZUVOG M throughout this leaflet.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any more questions, please ask your doctor or your pharmacist.
- This medicine has been prescribed for you personally and you should not pass it on to anyone else. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects get serious, or if you notice any side effects that are not listed in the leaflet, please tell your doctor or pharmacist.
In this leaflet:
1. What ZUVOG M Tablets are and what they are used for
2. What you need to know before you take ZUVOG M Tablets
3. How to take ZUVOG M Tablets
4. Possible side effects
5. How to store ZUVOG M Tablets
6. Contents of the pack and other information
1. What ZUVOG M Tablets are and What they are used for
ZUVOG M contains two active substances, voglibose and metformin hydrochloride. It is used as a second-line treatment for type II diabetes when diet, exercise, and either voglibose or metformin alone have not been enough to control blood glucose (sugar).
Voglibose works by blocking certain enzymes in your small intestine that break down complex sugars into simple sugars like glucose. By doing this, it slows down the absorption of sugar into your bloodstream after meals, helping to control blood sugar levels.
Insulin is a hormone that enables body tissues to take glucose from the blood and to use it for energy or for storage for future use. People with Type 2 diabetes do not make enough insulin in their pancreas or their body does not respond properly to the insulin it does make. This causes a build-up of glucose in the blood which can cause a number of serious long-term problems so it is important that you continue to take your medicine, even though you may not have any obvious symptoms. Metformin hydrochloride makes the body more sensitive to insulin and helps return to normal the way your body uses glucose.
2. What You Need to Know Before You Take Zuvog M Tablets
Do not take ZUVOG M if you:
- Are allergic to Metformin, Voglibose or any of the other ingredients of this medicine.
- Have inflammatory bowel disease, gastrointestinal obstruction, or conditions that may deteriorate due to increased gas formation.
- Have severe ketosis, diabetic coma or pre-coma, severe infection, or hepatic impairment.
- Have kidney disease or dysfunction.
- Are undergoing radiologic studies involving iodinated contrast materials.
- you have been treated for acute heart problems or have recently had a heart attack or have severe circulatory problems or breathing difficulties. This may lead to a lack in oxygen supply to tissue which can put you at risk for lactic acidosis
- you are a heavy drinker of alcohol.
- you are under 18 years of age.
Warnings and precautions
Risk of lactic acidosis
ZUVOG M Tablets may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration, liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease).
If any of the above apply to you, talk to your doctor for further instructions.
Stop taking ZUVOG M Tablets and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma.
Symptoms of lactic acidosis include:
- Vomiting
- stomach-ache (abdominal pain)
- muscle cramps
- a general feeling of not being well with severe tiredness
- difficulty in breathing
- reduced body temperature and heartbeat
Lactic acidosis is a medical emergency and must be treated in a hospital
Taking other medicines and ZUVOG M
- Antidiabetic Medications: Co-administration with sulfonylureas or insulin can enhance the hypoglycemic effect, increasing the risk of hypoglycemia. Close monitoring of blood glucose levels is recommended.
- Beta Blockers: These medications can mask the symptoms of hypoglycemia, making it harder to recognize low blood sugar levels.
- Keratolytics and MAOIs: These can enhance the hypoglycemic effect of Voglibose.
- Anti-lipidemic Agents: These can enhance the hypoglycemic effect of Voglibose.
- Sympathomimetics or Adrenergic Agonists: These can reduce the hypoglycemic effect of Voglibose.
- Adrenocorticotropic and Thyroid Hormones: These can reduce the hypoglycemic effect of Voglibose.
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
It is especially important to mention the following:
- Medicines which increase urine production (diuretics (water tablets) such as furosemide)
- medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as ibuprofen and celecoxib)
- certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists)
- Steroids such as prednisolone, mometasone, beclometasone.
- Sympathomimetic medicines including epinephrine and dopamine used to treat heart attacks and low blood pressure. Epinephrine is also included in some dental anaesthetics.
- Medicines that may change the amount of metformin in your blood, especially if you have reduced kidney function (such as verapamil, rifampicin, cimetidine, dolutegravir, ranolazine, trimethoprim, vandetanib, isavuconazole, crizotinib, olaparib).
ZUVOG M Tablets with alcohol
Avoid excessive alcohol intake while taking Metformin Hydrochloride Tablets since this may increase the risk of lactic acidosis.
Pregnancy
ZUVOG M is generally not recommended during pregnancy due to insufficient well-controlled studies in pregnant women. It should only be used if specifically advised by a healthcare professional in rare, carefully evaluated situations.
Breast-feeding
The use of ZUVOG M during breastfeeding is not well-studied, and there is limited data available on its safety for nursing mothers and infants. It is generally recommended to avoid ZUVOG M during breastfeeding unless absolutely necessary.
Driving and using machines
ZUVOG M does not usually affect your ability to drive or use machines. However, if you experience symptoms of hypoglycemia, such as dizziness or confusion, avoid these activities until you feel better.
3. How to Take Zuvog M Tablets
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
Dosage:
The initial recommended dose: one tablet of ZUVOG M 0.2 three times daily just before each meal.
If the effect is inadequate, the dose may be increased to one tablet of ZUVOG M 0.3 three times daily just before each meal.
Method of administration:
ZUVOG-M Tablet should be taken with food. Take it regularly at the same time each day to get the most benefit.
If you take more ZUVOG M Tablets than you should
Taking more Zuvog M Tablets than prescribed can lead to an overdose. While ZUVOG M is unlikely to cause hypoglycemia (low blood sugar) in overdose, it can cause significant gastrointestinal discomfort. Abdominal discomfort, diarrhea, Flatulence and Bloating may occur. Do not panic. Stay calm and seek medical advice immediately. Contact your doctor or go to the nearest hospital emergency department. Take the medicine packaging with you to show the healthcare professionals. Treatment is generally supportive and symptomatic, focusing on relieving gastrointestinal symptoms.
If you forget to take Zuvog M Tablets
If you forget to take a tablet, take one as soon as you remember, unless it is nearly time to take the next one. Never take two doses together. Take the remaining doses at the correct time.
If you stop taking Zuvog M Tablets
Take this medicine for as long as your doctor tells you to, as you may become unwell if you stop.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Voglibose
- Gastrointestinal Issues: Diarrhea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting, and heartburn.
- Abdominal Swelling and Increased Flatus: Symptoms similar to intestinal obstruction due to increased intestinal gas.
- Serious Hepatic Dysfunction: Jaundice, increased levels of liver enzymes (AST or ALT). One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported.
- Hyperammonemia: In patients with serious liver cirrhosis, voglibose may worsen hyperammonemia, leading to constipation and disturbance of consciousness.
- Hypoglycemia: When used in combination with other antidiabetic drugs.
Metformin:
- Common Adverse Reactions: Diarrhea, nausea/vomiting, flatulence, asthenia (weakness), indigestion, abdominal discomfort, headache, abnormal stools, hypoglycemia, myalgia (muscle pain), lightheadedness, dyspnea (difficulty breathing), nail disorder, rash, increased sweating, taste disorder, chest discomfort, chills, flu syndrome, flushing, and palpitations.
- Very Rare Adverse Effects: Liver function test abnormalities or hepatitis resolving upon discontinuation of metformin, and skin reactions such as erythema (redness of the skin), pruritus (itching), and urticaria (hives).
- Serious side effects: Lactic acidosis, characterized by symptoms such as deep or rapid breathing, persistent nausea, vomiting, and stomach pain.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly:
Website: www.zuventus.com and click the tab “Safety Reporting” located on the top t end of the home page.
Website link: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to Store Zuvog M Tablets
Do not use the tablets after the end of the expiry month (use-by date) shown on the product packaging.
Store below 30°C. Protected from moisture.
Keep this medicine out of the sight and reach of children
6. Contents of the Pack and Other Information
What ZUVOG M Tablets contain
The active substances are Voglibose and Metformin Hydrochloride.
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
Packaging:
ZUVOG M 0.2/0.3: A Blister strips of 10 tablets
For More Information About This Product
Zuvog M 0.2 mg Tablet
1.0 Generic Name
Voglibose and Metformin Hydrochloride (Prolonged-release) Tablets
2.0 Qualitative and quantitative composition
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
3.0 Dosage form and strength
Dosage Form: Tablets.
Dosage Strength: Voglibose 0.2 mg/ 0.3 mg and Metformin Hydrochloride 500 mg
(Prolonged-release)
4.0 Clinical particulars
4.1 Therapeutic Indication
As 2nd line treatment of type II Diabetes mellitus when diet, exercise and voglibose or metformin do not result in adequate glycemic control individually.
4.2 Posology and method of administration
Dosage should be individualised on the basis of both effectiveness and tolerance.
The initial recommended dose is one tablet of ZUVOG M 0.2 three times daily just before each meal.
In case of inadequate effect, the dose may be increased up to 1 tablet of ZUVOG M 0.3, three times daily just before each meal under close observation of the course of the disease.
4.3 Contraindications
- Renal disease or renal dysfunction (e.g., as suggested by serum creatinine levels 1.5 mg/dL [males], 1.4 mg/dL [females] or abnormal creatinine clearance), which may also result from conditions such as cardiovascular collapse (shock), acute myocardial infarction, and septicaemia.
- Hypersensitivity to metformin, voglibose or any of the components of this product.
- Acute or chronic metabolic acidosis, including diabetic ketoacidosis, with or without coma. Diabetic ketoacidosis should be treated with insulin.
- Severe infection, before and after surgery, serious trauma.
- Gastrointestinal obstruction or predisposed to it.
Patients undergoing radiologic studies involving intravascular administration of iodinated contrast materials should temporarily discontinue taking this combination, because use may result in acute alteration of renal function.
4.4 Special warnings and precautions for use
Loss of Control of Blood Glucose
When a patient stabilised on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of glycaemic control may occur. At such times, it may be necessary to withhold oral antidiabetic agents and temporarily administer insulin. This combinationmay be reinstituted after the acute episode is resolved.
Hypoxic States
Cardiovascular collapse (shock) from whatever cause, acute congestive heart failure, acute myocardial infarction and other conditions characterised by hypoxaemia have been associated with lactic acidosis and may also cause prerenalazotaemia. When such events occur in patients on ZUVOG M, the drug should be promptly discontinued.
Surgical Procedures
ZUVOG M should be temporarily suspended for any surgical procedure (except minor procedures not associated with restricted intake of food and fluids) and should not be restarted until the patient’s oral intake has resumed and renal function has been evaluated as normal.
Alcohol Intake
Alcohol is known to potentiate the effect of metformin on lactate metabolism. Patients, therefore, should be warned against excessive alcohol intake, acute or chronic, while receiving ZUVOG M.
Change in Clinical Status of Patients with Previously Controlled Type 2 Diabetes
A patient with type 2 diabetes with previously well controlled type 2 diabetes who develops laboratory abnormalities or clinical illness (especially vague and poorly defined illness) should be evaluated promptly for evidence of ketoacidosis or lactic acidosis. Evaluation should include serum electrolytes and ketones, blood glucose and, if indicated, blood pH, lactate, pyruvate, and metformin levels. If acidosis of either form occurs, ZUVOG M must be stopped immediately and other appropriate corrective measures initiated.
Hypoglycaemia
Hypoglycaemia does not occur in patients receiving ZUVOG M alone under usual circumstances of use, but could occur when caloric intake is deficient, when strenuous exercise is not compensated by caloric supplementation, or during concomitant use with other glucose-lowering agents (such as sulphonylureas and insulin) or ethanol. Elderly, debilitated, or malnourished patients and those with adrenal or pituitary insufficiency or alcohol intoxication are particularly susceptible to hypoglycaemic effects. Hypoglycaemia may be difficult to recognize in the elderly, and in people who are taking beta-adrenergic blocking drugs. Patients should be instructed and explained to recognize hypoglycaemic symptoms and its management.
Vitamin B12
In controlled clinical trials of metformin of 29 weeks duration, a decrease to subnormal levels of previously normal serum vitamin B12 levels, without clinical manifestations, was observed in approximately 7% of patients. Such decrease, possibly due to interference with B12 absorption from the B12 intrinsic factor complex, however is very rarely associated with anaemia and appears to be rapidly reversible with discontinuation of metformin or vitamin B12 supplementation. Measurement of haematologic parameters on an annual basis is advised in patients on metformin and any apparent abnormalities should be appropriately investigated and managed.
Certain individuals (those with inadequate vitamin B12 or calcium intake or absorption) appear to be predisposed to developing subnormal vitamin B12 levels. In these patients, routine serum vitamin B12 measurements at 2- or 3- year intervals may be useful.
Macrovascular Outcomes
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with voglibose, metformin or any other anti-diabetic drug.
Lactic Acidosis
Lactic acidosis is a rare, but serious, metabolic complication that can occur due to metformin accumulation during treatment with metformin; when it occurs, it is fatal in approximately 50% of cases. Lactic acidosis may also occur in association with a number of pathophysiologic conditions, including diabetes mellitus, and whenever there is significant tissue hypo perfusion and hypoxaemia. Lactic acidosis is characterised by elevated blood lactate levels (>5 mmol/L), decreased blood pH, electrolyte disturbances with an increased anion gap, and an increased lactate/pyruvate ratio. When metformin is implicated as the cause of lactic acidosis, metformin plasma levels >5 μg/mL are generally found.
The reported incidence of lactic acidosis in patients receiving metformin hydrochloride is very low. Patients with congestive heart failure requiring pharmacologic management, in particular those with unstable or acute congestive heart failure who are at risk of hypo perfusion and hypoxaemia, are at increased risk of lactic acidosis. The risk of lactic acidosis increases with the degree of renal dysfunction and the patient’s age. The risk of lactic acidosis may, therefore, be significantly decreased by regular monitoring of renal function in patients taking metformin and by use of the minimum effective dose of metformin. In particular, treatment of the elderly should be accompanied by careful monitoring of renal function. Metformin treatment should not be initiated in patients ≥80 years of age unless measurement of creatinine clearance demonstrates that renal function is not reduced, as these patients are more susceptible to developing lactic acidosis. In addition, metformin should be promptly withheld in the presence of any condition associated with hypoxaemia, dehydration, or sepsis. Because impaired hepatic function may significantly limit the ability to clear lactate, metformin should generally be avoided in patients with clinical or laboratory evidence of hepatic disease. Patients should be cautioned against excessive alcohol intake, either acute or chronic, when taking metformin, since alcohol potentiates the effects of metformin hydrochloride on lactate metabolism. In addition, metformin should be temporarily discontinued prior to any intravascular radiocontrast study and for any surgical procedure.
The onset of lactic acidosis often is subtle, and accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, increasing somnolence, and nonspecific abdominal distress. There may be associated hypothermia, hypotension, and resistant bradyarrhythmias with more marked acidosis. The patient and the patient’s physician must be aware of the possible importance of such symptoms and the patient should be instructed to notify the physician immediately if they occur. Metformin should be withdrawn until the situation is clarified. Serum electrolytes, ketones, blood glucose, and if indicated, blood pH, lactate levels, and even blood metformin levels may be useful. Once a patient is stabilised on any dose level of metformin, gastrointestinal symptoms, which are common during initiation of therapy, are unlikely to be drug related. Later occurrence of gastrointestinal symptoms could be due to lactic acidosis or other serious disease.
Levels of fasting venous plasma lactate above the upper limit of normal but less than 5 mmol/L in patients taking metformin do not necessarily indicate impending lactic acidosis and may be explainable by other mechanisms, such as poorly controlled diabetes or obesity, vigorous physical activity, or technical problems in sample handling.
Lactic acidosis should be suspected in any diabetes patient with metabolic acidosis lacking evidence of ketoacidosis (ketonuria and ketonemia).
Lactic acidosis is a medical emergency that must be treated in a hospital setting. In a patient with lactic acidosis who is taking metformin, the drug should be discontinued immediately and general supportive measures promptly instituted. Because metformin hydrochloride is dialyzable (with a clearance of up to 170 mL/min under good hemodynamic conditions), prompt haemodialysis is recommended to correct the acidosis and remove the accumulated metformin. Such management often results in prompt reversal of symptoms and recovery.
4.5 Drugs interactions
Cationic Drugs
Certain medications used concomitantly with metformin may increase the risk of lactic acidosis. Cationic drugs that are eliminated by renal tubular secretions (e.g: amiloride, cimetidine, digoxin, morphine, procainamide, quinidine, quinine, ranitidine, triamterene, trimethoprim or vancomycin) may decrease metformin elimination by competing for common renal tubular transport systems. Hence, careful patient monitoring and dose adjustment of ZUVOG M and/or interfering drug is recommended in patients who are taking cationic medications that are excreted via the proximal renal tubular secretory system.
Furosemide
A single-dose, metformin-furosemide drug interaction study in healthy subjects demonstrated that pharmacokinetic parameters of both compounds were affected by co-administration. Furosemide increased the metformin plasma and blood Cmax by 22% and blood AUC by 15%, without any significant change in metformin renal clearance. When administered with metformin, the Cmax and AUC of furosemide were 31% and 12% smaller, respectively, than when administered alone, and the terminal half-life was decreased by 32%, without any significant change in furosemide renal clearance. No information is available about the interaction of metformin and furosemide when co-administered chronically.
Nifedipine
Nifedipine appears to enhance the absorption of metformin, it increases plasma metformin Cmax and AUC by 20% and 9% respectively and increases the amount of metformin excreted in the urine. Metformin has minimal effects on nifedipine.
Danazol
If the use of this active substance cannot be avoided, warn the patients and emphasise the importance of urine and blood glucose monitoring. It may be necessary to adjust the dose of voglibose and metformin during and after treatment with danazol.
Salicylates
If salicylates are administered or discontinued in patients receiving oral antidiabetic agents, patients should be monitored for hypoglycaemia or loss of blood glucose control.
Thiazide
Interactions between thiazide diuretics and oral antidiabetic agent’s decreases insulin sensitivity thereby leading to glucose intolerance and hyperglycaemia, thus leading to a loss of diabetic control. Hence diabetic patients should be monitored closely.
Other
Concomitant administration of angiotensin enzyme inhibitors (captopril, enalapril), other antidiabetic drugs (insulin, acarbose) beta-blockers, fluconazole, histamine (H2) receptor antagonist, monoamine oxidase inhibitors (MAOIs), sulphonamides and non-steroidal anti-inflammatory agents increase sensitivity to insulin and potentiation of blood glucose lowering effect and thus, in some instances, hypoglycaemia may occur . Certain drugs tend to produce hyperglycaemia and may lead to loss of glycaemic control. These drugs include corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel blocking drugs, quinolones and isoniazid. Patients receiving these drugs should be closely monitored for loss of diabetic control when therapy is instituted or discontinued. Dosage of the oral antidiabetic agents may need to be reduced.
Drugs Enhancing (beta-blockers, salicylic acid preparations, MAOs, fibrate derivatives) or Diminishing (epinephrine, adrenocortical hormone, thyroid hormone etc.) the Hypoglycaemic Action of Anti-Diabetic Drugs
When ZUVOG M is administered concomitantly with drugs that enhance or diminish the hypoglycaemic action of anti-diabetic drugs, caution should be taken as this might additionally delay the action of voglibose on the absorption of carbohydrates.
Warfarin
Metformin and voglibose do not affect the pharmacokinetics of warfarin, hence ZUVOG M can be safely administered along with warfarin.
4.6 Use in special populations
Pregnant Women
Recent information suggests that abnormal blood glucose levels during pregnancy are associated with the higher incidence of congenital abnormalities. There are no adequate and well-controlled studies in pregnant women with metformin. Metformin was not teratogenic in rats and rabbits at doses up to 600 mg/kg/day. The safety and effectiveness of voglibose in pregnant women has not been established. Animal studies have shown that voglibose is transferred to the foetus. ZUVOG M should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus. Most experts suggest insulin be used to maintain the blood glucose levels as close to normal as possible.
Lactating Women
Studies in lactating rats show that metformin is excreted into milk and reaches levels comparable to those in plasma. Similar studies have not been conducted on nursing mothers. Animal studies have shown a suppressive action of voglibose on body weight increase in newborns, mainly due to suppression of milk production resulting from inhibition of carbohydrate absorption in mother animals. Nursing should be discontinued if ZUVOG M has to be administered. If the use of this combination is discontinued, and if the diet alone is inadequate for controlling blood glucose, insulin therapy should be considered.
Paediatric Patients
The safety and effectiveness of metformin for the treatment of type 2 diabetes have been established in paediatric patients ages 10 to 16 years (studies have not been conducted in paediatric patients below the age of 10 years). The safety and effectiveness of voglibose in children has not been established. Hence ZUVOG M should not be considered in this population.
Geriatric
Metformin is known to be excreted through the kidneys and because risk of serious adverse reactions to the drug is greater in patients with impaired renal function, ZUVOG M should be used only in patients with normal renal function. Because aging is associated with reduced renal function, the use of ZUVOG M should be with caution as age increases. Care should be taken in the dose selection and regular renal function be monitored. The administration of ZUVOG M should be initiated at a lower dose in elderly patients and generally, should not be titrated to the maximum dose. The combination should be carefully administered under close observation of the course of disease conditions, with careful attention to the blood sugar level and the onset of gastrointestinal symptoms.
Renal Impairment Patients
Metformin is known to be substantially excreted by the kidney and the risk of metformin accumulation and lactic acidosis increases with the degree of impairment of renal function. Thus, patients with serum creatinine levels above the upper limit of normal for their age should not receive ZUVOG M. In patients with advanced age, ZUVOG M should be carefully titrated to establish the minimum dose for adequate glycaemic effect, because aging is associated with reduced renal function. In elderly patients, particularly those ≥80 years of age, renal function should be monitored regularly and, generally, ZUVOG M should not be titrated to the maximum dose. Before initiation of ZUVOG M and at least annually thereafter, renal function should be assessed and verified as normal. In patients in whom development of renal dysfunction is anticipated, renal function should be assessed more frequently and ZUVOG M discontinued if evidence of renal impairment is present.
4.7 Effects on ability to drive and use machines
This combination containing voglibose and metformin does not cause hypoglycaemia and therefore has no effect on the ability to drive or to use machines. However, patients should be alerted to the risk of hypoglycaemia when ZUVOG M is used in combination with other antidiabetic agents (e.g. sulphonylureas, insulin, or meglinitides).
4.8 Undesirable effects
Voglibose
The gastrointestinal adverse effects like diarrhoea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting or heartburn may occur with the use of voglibose. Abdominal swelling, increased flatus, and intestinal obstruction like symptoms due to an increase in intestinal gas, etc. may occur. Serious hepatic dysfunction accompanied with jaundice, increased aspartate aminotransferase (AST) or alanine aminotransferase (ALT), etc. may also occur. One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported; a causal relationship appears likely. When voglibose is administered to the patients with serious liver cirrhosis, hyperammonia may worsen with the development of constipation, etc., followed by disturbance of consciousness. When voglibose is used in combination with other antidiabetic drugs, hypoglycaemia may occur.
Metformin
The most common adverse reactions of metformin hydrochloride are diarrhoea, nausea/vomiting, flatulence, asthenia, indigestion, abdominal discomfort, headache, abnormal stools, hypoglycaemia, myalgia, lightheaded, dyspnea, nail disorder, rash, sweating increased, taste disorder, chest discomfort, chills, flu syndrome, flushing and palpitation. The very rare adverse effects include isolated reports of liver function tests, abnormalities or hepatitis resolving upon metformin discontinuation and skin reactions such as erythema, pruritus, urticaria.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
4.9 Overdose
Overdose of metformin hydrochloride has occurred, including ingestion of amounts greater than 50 grams. Hypoglycaemia was reported in approximately 10% of cases, but no causal association with metformin hydrochloride has been established. Lactic acidosis has been reported in approximately 32% of metformin overdose cases. Metformin is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions. Therefore, haemodialysis may be useful for removal of accumulated drug from patients in whom metformin overdosage is suspected.
Voglibose competitively and reversibly inhibits the alpha-glucosidase enzymes in the brush border in the small intestine, which delays the hydrolysis of complex carbohydrates. It appears unlikely to produce hypoglycaemia in overdose, but abdominal discomfort and diarrhea may occur.
5.0 Pharmacological properties
5.1 Pharmacodynamic/ Mechanism of Action
Voglibose
Voglibose is an alpha glucosidase inhibitor which inhibits the activity of alpha glucosidases that catalyse the decomposition of disaccharides into monosaccharides in the intestine, thereby delaying the digestion and absorption of carbohydrates, resulting in improvement of postprandial hyperglycaemia.
Metformin Hydrochloride
Metformin improves glucose tolerance in patients with type 2 diabetes (NIDDM), lowering both basal and postprandial plasma glucose. Metformin is not chemically or pharmacologically related to sulphonylureas, thiazolidinediones, or alpha-glucosidase inhibitors. Metformin decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. Unlike sulphonylureas, metformin does not produce hypoglycaemia in either patient with type 2 diabetes or normal subjects and does not cause hyperinsulinaemia. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may actually decrease.
5.2 Pharmacokinetic properties
Voglibose
Voglibose is poorly absorbed after oral doses. Plasma concentrations after oral doses have usually been undetectable. Following repeated administration to healthy subjects (n=6) in a single dose of 0.2 mg, 3 times a day, for 7 consecutive days, voglibose was not detected in plasma or urine. Similarly, when voglibose was administered to healthy male adults (n=10) as a single dose of 2 mg, voglibose was not detected in plasma or urine. After ingestion of voglibose, the majority of active unchanged drug remains in the lumen of the gastrointestinal tract to exert its pharmacological activity. Voglibose is metabolized by intestinal enzymes and by the microbial flora. Voglibose is mainly excreted in the feces.
Metformin Hydrochloride
The absolute bioavailability of a metformin 500-mg tablet given under fasting conditions is approximately 50-60%. Studies using single oral doses of metformin 500 mg to 1500 mg, and 850 mg to 2550 mg, indicate that there is a lack of dose proportionality with increasing doses, which is due to decreased absorption rather than an alteration in elimination. Food decreases the extent of and slightly delays the absorption of metformin, as shown by approximately a 40% lower mean peak plasma concentration (Cmax), a 25% lower area under the plasma concentration versus time curve (AUC), and a 35-minute prolongation of time to peak plasma concentration (Tmax) following administration of a single 850-mg tablet of metformin with food, compared to the same tablet strength administered fasting. The clinical relevance of these decreases is unknown.
The apparent volume of distribution (V/F) of metformin following single oral doses of metformin immediate-release 850 mg averaged 654 ± 358 L. Metformin is negligibly bound to plasma proteins, in contrast to sulphonylureas, which are more than 90% protein bound. Metformin partitions into erythrocytes, most likely as a function of time. At usual clinical doses and dosing schedules of metforminimmediate-release, steady state plasma concentrations of metformin are reached within 24-48 hours and are generally <1 µg/mL. During controlled clinical trials of immediate-release metformin, maximum metformin plasma levels did not exceed 5 µg/mL, even at maximum doses.
Intravenous single-dose studies in normal subjects demonstrate that metformin is excreted unchanged in the urine and does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion. Renal clearance of metformin is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of metformin elimination. Following oral administration, approximately 90% of the absorbed drug is eliminated via the renal route within the first 24 hours, with a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Voglibose
A study demonstrated that voglibose can potentiate CCl4 (carbon tetrachloride) and APAP (acetaminophen) hepatotoxicity in rats by inducing hepatic CYP2E1.
Metformin
Long-term carcinogenicity studies have been performed in rats (dosing duration of 104 weeks) and mice (dosing duration of 91 weeks) at doses up to and including 900 mg/kg/day and 1500 mg/kg/day, respectively. These doses are both approximately 3 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons. No evidence of carcinogenicity with metformin was found in either male or female mice. Similarly, there was no tumorigenic potential observed with metformin in male rats. There was, however, an increased incidence of benign stromal uterine polyps in female rats treated with 900 mg/kg/day.
There was no evidence of a mutagenic potential of metformin in the following in vitro tests: Ames test (S. typhimurium), gene mutation test (mouse lymphoma cells), or chromosomal aberrations test (human lymphocytes). Results in the in vivo mouse micronucleus test were also negative.
Fertility of male or female rats was unaffected by metformin when administered at doses as high as 600 mg/kg/day, which is approximately 2 times the maximum recommended human daily dose of 2550 mg based on body surface area comparisons.
7.0 Description
Zuvog M contains two oral antihyperglycemic drugs, voglibose and metformin hydrochloride, used in the management of type 2 diabetes mellitus.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
Zuvog M 0.2 / 0.3 - A blister strip of 10 tablets
8.4 Storage and handing instructions
Store below 30°C. Protect from moisture.
Keep out of reach of children.
9.0 Patient Counselling Information
- Tablet should be swallowed whole & not to be broken, crushed or chewed.
- Take this medicine exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting your doctor.
- Tell your doctor immediately if you experience any deep or rapid breathing, persistent nausea, vomiting, and stomach pain as ZUVOG M Tablet may cause a rare but serious condition called lactic acidosis, which is an excess of lactic acid in the blood.
- Pregnant women and breastfeeding mothers should not use this medicine without doctor consultation.
- Don’t medicine for the treatment of diabetic ketoacidosis or diabetic pre-coma.
- This medicine is not advisable for use in children.
- Avoid use of this medicine during severe infection or before and after surgery or in serious trauma cases.
- This tablet may cause hypoglycemia when co-administered with oral sulfonylureas and insulin.
12.0 Date of revision
11/10/2024
The name of your medicine is ZUVOG M. We refer to them as ZUVOG M TABLETS or ZUVOG M throughout this leaflet.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any more questions, please ask your doctor or your pharmacist.
- This medicine has been prescribed for you personally and you should not pass it on to anyone else. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects get serious, or if you notice any side effects that are not listed in the leaflet, please tell your doctor or pharmacist.
In this leaflet:
1. What ZUVOG M Tablets are and what they are used for
2. What you need to know before you take ZUVOG M Tablets
3. How to take ZUVOG M Tablets
4. Possible side effects
5. How to store ZUVOG M Tablets
6. Contents of the pack and other information
1. What ZUVOG M Tablets are and What they are used for
ZUVOG M contains two active substances, voglibose and metformin hydrochloride. It is used as a second-line treatment for type II diabetes when diet, exercise, and either voglibose or metformin alone have not been enough to control blood glucose (sugar).
Voglibose works by blocking certain enzymes in your small intestine that break down complex sugars into simple sugars like glucose. By doing this, it slows down the absorption of sugar into your bloodstream after meals, helping to control blood sugar levels.
Insulin is a hormone that enables body tissues to take glucose from the blood and to use it for energy or for storage for future use. People with Type 2 diabetes do not make enough insulin in their pancreas or their body does not respond properly to the insulin it does make. This causes a build-up of glucose in the blood which can cause a number of serious long-term problems so it is important that you continue to take your medicine, even though you may not have any obvious symptoms. Metformin hydrochloride makes the body more sensitive to insulin and helps return to normal the way your body uses glucose.
2. What You Need to Know Before You Take Zuvog M Tablets
Do not take ZUVOG M if you:
- Are allergic to Metformin, Voglibose or any of the other ingredients of this medicine.
- Have inflammatory bowel disease, gastrointestinal obstruction, or conditions that may deteriorate due to increased gas formation.
- Have severe ketosis, diabetic coma or pre-coma, severe infection, or hepatic impairment.
- Have kidney disease or dysfunction.
- Are undergoing radiologic studies involving iodinated contrast materials.
- you have been treated for acute heart problems or have recently had a heart attack or have severe circulatory problems or breathing difficulties. This may lead to a lack in oxygen supply to tissue which can put you at risk for lactic acidosis
- you are a heavy drinker of alcohol.
- you are under 18 years of age.
Warnings and precautions
Risk of lactic acidosis
ZUVOG M Tablets may cause a very rare, but very serious side effect called lactic acidosis, particularly if your kidneys are not working properly. The risk of developing lactic acidosis is also increased with uncontrolled diabetes, serious infections, prolonged fasting or alcohol intake, dehydration, liver problems and any medical conditions in which a part of the body has a reduced supply of oxygen (such as acute severe heart disease).
If any of the above apply to you, talk to your doctor for further instructions.
Stop taking ZUVOG M Tablets and contact a doctor or the nearest hospital immediately if you experience some of the symptoms of lactic acidosis, as this condition may lead to coma.
Symptoms of lactic acidosis include:
- Vomiting
- stomach-ache (abdominal pain)
- muscle cramps
- a general feeling of not being well with severe tiredness
- difficulty in breathing
- reduced body temperature and heartbeat
Lactic acidosis is a medical emergency and must be treated in a hospital
Taking other medicines and ZUVOG M
- Antidiabetic Medications: Co-administration with sulfonylureas or insulin can enhance the hypoglycemic effect, increasing the risk of hypoglycemia. Close monitoring of blood glucose levels is recommended.
- Beta Blockers: These medications can mask the symptoms of hypoglycemia, making it harder to recognize low blood sugar levels.
- Keratolytics and MAOIs: These can enhance the hypoglycemic effect of Voglibose.
- Anti-lipidemic Agents: These can enhance the hypoglycemic effect of Voglibose.
- Sympathomimetics or Adrenergic Agonists: These can reduce the hypoglycemic effect of Voglibose.
- Adrenocorticotropic and Thyroid Hormones: These can reduce the hypoglycemic effect of Voglibose.
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
It is especially important to mention the following:
- Medicines which increase urine production (diuretics (water tablets) such as furosemide)
- medicines used to treat pain and inflammation (NSAID and COX-2-inhibitors, such as ibuprofen and celecoxib)
- certain medicines for the treatment of high blood pressure (ACE inhibitors and angiotensin II receptor antagonists)
- Steroids such as prednisolone, mometasone, beclometasone.
- Sympathomimetic medicines including epinephrine and dopamine used to treat heart attacks and low blood pressure. Epinephrine is also included in some dental anaesthetics.
- Medicines that may change the amount of metformin in your blood, especially if you have reduced kidney function (such as verapamil, rifampicin, cimetidine, dolutegravir, ranolazine, trimethoprim, vandetanib, isavuconazole, crizotinib, olaparib).
ZUVOG M Tablets with alcohol
Avoid excessive alcohol intake while taking Metformin Hydrochloride Tablets since this may increase the risk of lactic acidosis.
Pregnancy
ZUVOG M is generally not recommended during pregnancy due to insufficient well-controlled studies in pregnant women. It should only be used if specifically advised by a healthcare professional in rare, carefully evaluated situations.
Breast-feeding
The use of ZUVOG M during breastfeeding is not well-studied, and there is limited data available on its safety for nursing mothers and infants. It is generally recommended to avoid ZUVOG M during breastfeeding unless absolutely necessary.
Driving and using machines
ZUVOG M does not usually affect your ability to drive or use machines. However, if you experience symptoms of hypoglycemia, such as dizziness or confusion, avoid these activities until you feel better.
3. How to Take Zuvog M Tablets
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
Dosage:
The initial recommended dose: one tablet of ZUVOG M 0.2 three times daily just before each meal.
If the effect is inadequate, the dose may be increased to one tablet of ZUVOG M 0.3 three times daily just before each meal.
Method of administration:
ZUVOG-M Tablet should be taken with food. Take it regularly at the same time each day to get the most benefit.
If you take more ZUVOG M Tablets than you should
Taking more Zuvog M Tablets than prescribed can lead to an overdose. While ZUVOG M is unlikely to cause hypoglycemia (low blood sugar) in overdose, it can cause significant gastrointestinal discomfort. Abdominal discomfort, diarrhea, Flatulence and Bloating may occur. Do not panic. Stay calm and seek medical advice immediately. Contact your doctor or go to the nearest hospital emergency department. Take the medicine packaging with you to show the healthcare professionals. Treatment is generally supportive and symptomatic, focusing on relieving gastrointestinal symptoms.
If you forget to take Zuvog M Tablets
If you forget to take a tablet, take one as soon as you remember, unless it is nearly time to take the next one. Never take two doses together. Take the remaining doses at the correct time.
If you stop taking Zuvog M Tablets
Take this medicine for as long as your doctor tells you to, as you may become unwell if you stop.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Voglibose
- Gastrointestinal Issues: Diarrhea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting, and heartburn.
- Abdominal Swelling and Increased Flatus: Symptoms similar to intestinal obstruction due to increased intestinal gas.
- Serious Hepatic Dysfunction: Jaundice, increased levels of liver enzymes (AST or ALT). One case of hepatitis with severe cholestasis attributed to voglibose hypersensitivity has been reported.
- Hyperammonemia: In patients with serious liver cirrhosis, voglibose may worsen hyperammonemia, leading to constipation and disturbance of consciousness.
- Hypoglycemia: When used in combination with other antidiabetic drugs.
Metformin:
- Common Adverse Reactions: Diarrhea, nausea/vomiting, flatulence, asthenia (weakness), indigestion, abdominal discomfort, headache, abnormal stools, hypoglycemia, myalgia (muscle pain), lightheadedness, dyspnea (difficulty breathing), nail disorder, rash, increased sweating, taste disorder, chest discomfort, chills, flu syndrome, flushing, and palpitations.
- Very Rare Adverse Effects: Liver function test abnormalities or hepatitis resolving upon discontinuation of metformin, and skin reactions such as erythema (redness of the skin), pruritus (itching), and urticaria (hives).
- Serious side effects: Lactic acidosis, characterized by symptoms such as deep or rapid breathing, persistent nausea, vomiting, and stomach pain.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly:
Website: www.zuventus.com and click the tab “Safety Reporting” located on the top t end of the home page.
Website link: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to Store Zuvog M Tablets
Do not use the tablets after the end of the expiry month (use-by date) shown on the product packaging.
Store below 30°C. Protected from moisture.
Keep this medicine out of the sight and reach of children
6. Contents of the Pack and Other Information
What ZUVOG M Tablets contain
The active substances are Voglibose and Metformin Hydrochloride.
Each uncoated bilayered tablet contains:
Voglibose IP………………………0.2 mg/ 0.3 mg
Metformin Hydrochloride IP……...500 mg
(As Prolonged-release form)
Excipients……………………………. q.s.
Packaging:
ZUVOG M 0.2/0.3: A Blister strips of 10 tablets
For More Information About This Product
Zuvog 0.3 mg Tablet
1.0 Generic Name
Voglibose Tablets 0.2mg / 0.3mg
2.0 Qualitative and quantitative composition
Each uncoated tablet contains:
Voglibose……………….0.2mg / 0.3mg
Excipients q.s.
3.0 Dosage form and strength
Dosage Form: Tablets.
Dosage Strength: Voglibose 0.2 mg and 0.3 mg per tablet.
4.0 Clinical particulars
4.1 Therapeutic Indication
For improvement of post prandial hyperglycaemia in diabetic mellitus only when diet and/or exercise or oral hypoglycemic drugs or insulin preparations in addition to diet and /or exercise do not result adequate glycemic control.
4.2 Posology and method of administration
ZUVOG is orally administered in a single dose of 0.2 mg three times daily just before or with each meal.
ZUVOG should be administered in conjunction with diet treatment or diet plus a sulphonylurea.
In case of inadequate effect, the single dose may be increased up to 0.3 mg under close observation of the course of the disease.
4.3 Contraindication
- Hypersensitivity to voglibose or to any component of the formulation.
- Diabetic ketoacidosis, diabetic pre-coma.
- Severe infection, before and after surgery, serious trauma.
- Gastrointestinal obstruction or predisposed to it
4.4 Special warnings and precautions for use
Hypoglycaemia
While on voglibose tablets, patients should be instructed and explained to recognize hypoglycaemic symptoms and its management.
Loss of Control of Blood Glucose
When diabetes patients are exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of control of blood glucose may occur. At such times, temporary insulin therapy may be necessary.
4.5 Drugs interactions
Anti-Diabetic Drugs: When voglibose is used in combination with derivative(s) of sulfonylamide, sulfonylurea or biguanide, or with insulin, hypoglycemic symptoms may occur. Therefore, when used in combination with any of these drugs, care should be taken, such as to initiate therapy with lower dosage.
Drugs Affecting Glycemic Control: When voglibose is administered concomitantly with drugs that enhance or diminish the hypoglycemic action of antidiabetic drugs, caution should be taken as this might additionally delay the action of voglibose on the absorption of carbohydrates. Examples of drugs enhancing the hypoglycemic action of antidiabetic drugs include alpha-blockers, salicylic acid preparations, monoamine oxidase inhibitors, and fibrate derivatives. Examples of drugs diminishing the hypoglycemic action of antidiabetic drugs include epinephrine, adrenocortical hormone, and thyroid hormone.
Warfarin: Voglibose does not affect the pharmacokinetics of warfarin; hence, it can be safely administered along with warfarin.
4.6 Use in special populations
Pregnant Women
Pregnancy Category B. The safety of voglibose in pregnancy has not been established. Animal studies do not indicate harmful effects with respect to pregnancy, embryonal or fetal development, parturition or post-natal development. However, no adequate and well controlled studies have been done in pregnant women. Therefore, voglibose should be given to pregnant women only when the potential benefits to the mother outweigh the possible hazards to the fetus.
Lactating Women
Animal studies (e.g., rats) have revealed a suppressive action of voglibose on body weight increase in newborns probably due to suppression of milk production due to reduced carbohydrate absorption. Although the levels of voglibose reached in human milk are exceedingly low, it is recommended that voglibose may not be administered to lactating women. When the administration of voglibose is unavoidable, nursing should be discontinued.
Paediatric Patients
The safety and effectiveness of voglibose in children has not been established. Thus, Zuvog Tablets are not recommended for use in paediatric patients.
Geriatric
Patients Since elderly patients generally have a physiological hypofunction, it is desirable to initiate therapy with lower dosage. Moreover, this drug should be carefully administered under close observation, throughout the course of the disease, with careful attention to the blood sugar level and the onset of gastrointestinal symptoms.
Renal Impairment Patients
Voglibose is poorly absorbed after oral doses and renal excretion is negligible. Thus, in general, no dosage adjustment is required in patients with renal dysfunction.
Effects on ability to drive and use machines
4.7 Effects on ability to drive and use machines
With voglibose, effect on ability to drive a vehicle or operating machinery has not been reported.
4.8 Undesirable effects
- Gastrointestinal: Gastrointestinal adverse events such as diarrhoea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting, or heartburn may occur with the use of voglibose. Also, abdominal distention, increased flatus, and intestinal obstruction like symptoms due to an increase in intestinal gas may occur with use of voglibose.
- Hypersensitivity: Rash and pruritus may rarely occur. In such cases, voglibose should be discontinued immediately.
- Hepatic: When voglibose is administered to patients with liver cirrhosis, hyperammonia may worsen with the development of constipation followed by disturbance of consciousness.
- Laboratory Tests: Elevation of SGOT (serum glutamate oxaloacetate), SGPT (serum glutamate pyruvate transaminase), LDH (lactate dehydrogenase), alpha-GPT (alpha-glutamate pyruvate transaminase) or alkaline phosphatase may infrequently occur.
- Hypoglycemia: When voglibose is used in combination with other antidiabetic drugs, hypoglycemia may occur (0.1% to <5%).
- Psychoneurologic: Headache may rarely occur.
- Hematologic: Anemia, thrombocytopenia, and leucopoenia may rarely occur.
- Others: Numbness, edema of face, blurred vision, hot flushes, malaise, weakness, hyperkalemia, increased serum amylase, decreased HDL-cholesterol, diaphoresis, or alopecia may occur rarely with the use of voglibose.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to:medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
4.9 Overdose
Voglibose is unlikely to produce hypoglycemia in overdose, but abdominal discomfort and diarrhoea may occur. If overdose occurs, supportive and symptomatic treatment should be provided.
5.0 Pharmacological properties
5.1 Mechanism of Action
Voglibose competitively and reversibly inhibits the alpha glucosidase enzymes (e.g., glucoamylase, sucrase, maltase, and isomaltase) in the brush border of the small intestine. Alpha-glucosidase enzymes are essential for hydrolysis/decomposition of complex carbohydrates (starch, dextrin, polysaccharides, and disaccharides) into simpler carbohydrates (such as glucose/dextrose or fructose). Inhibition of these enzymes leads to delay in the absorption of glucose into the bloodstream resulting in improvement of post-prandial hyperglycemia.
5.2 Pharmacodynamic properties
Voglibose exerts its activity in the intestinal tract. Alpha glucosidase enzyme normally converts complex carbohydrates into simple monosaccharides (glucose) which can be absorbed through the intestine.
The action of voglibose depends on an inhibition of intestinal enzymes (alpha-glucosidase) involved in the degradation of ingested disaccharides, oligosaccharides, and polysaccharides into monosaccharides. Inhibition of these enzyme systems reduces/delays the rate of digestion of complex carbohydrates. As there is delay in digestion of complex carbohydrates, monosaccharides release slowly and hence absorbed more slowly into the blood i.e., less glucose absorption from intestine into the blood circulation. Voglibose, thus dose dependently reduces the postprandial rise in blood glucose level.
Although voglibose reduces the impact of complex carbohydrates on blood sugar level, it does not affect/inhibit absorption of glucose from the intestine.
Alpha-glucosidase inhibitors such as voglibose do not stimulate insulin release and therefore do not result in hypoglycemia. Voglibose improves post-prandial hyperglycemia and thereby lowers abnormally high levels of glycosylated hemoglobin. Voglibose is highly useful in elderly patients or in patients with predominantly post-prandial hyperglycemia.
5.3 Pharmacokinetic properties
Absorption: Voglibose is poorly absorbed after oral doses. Plasma concentrations after oral doses have usually been undetectable. Following repeated administration to healthy subjects (n=6) in a single dose of 0.2 mg, 3 times a day, for 7 consecutive days, voglibose was not detected in plasma or urine. Similarly, when voglibose was administered to healthy male adults (n=10) as a single dose of 2 mg, voglibose was not detected in plasma or urine.
Distribution: After ingestion of voglibose, the majority of active unchanged drug remains in the lumen of the gastrointestinal tract to exert its pharmacological activity.
Metabolism: Voglibose is metabolized by intestinal enzymes and by the microbial flora. Excretion: Voglibose is mainly excreted in the feces.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
A study demonstrated that voglibose can potentiate CCl4 (carbon tetrachloride) and APAP (acetaminophen) hepatotoxicity in rats by inducing hepatic CYP2E1.
7.0 Description
Voglibose is an alpha-glucosidase inhibitor used to manage postprandial blood glucose levels in patients with type 2 diabetes mellitus.
Chemical name: (1S, 2S, 3R, 4S, 5S)-5-(1,3-dihydroxypropan-2-ylamino)-1-(hydroxymethyl) cyclohexane-1,2,3,4-tetrol.
Molecular formula: C10H21NO7
Molecular mass: 267.28 g/mol.
Structural formula:

8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
Zuvog 0.2 / 0. 3: A blister strip of 10 tablets
8.4 Storage and handing instructions
Store below 25°C. Protected from moisture.
9.0 Patient Counselling Information
- Take this medicine exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting your doctor.
- Pregnant women and breastfeeding mothers should not use this medicine without doctor consultation.
- Don’t medicine for the treatment of diabetic ketoacidosis or diabetic pre-coma.
- This medicine is not advisable for use in children.
- Avoid use of this medicine during severe infection or before and after surgery or in serious trauma cases.
12.0 Date of revision
10/10/2024
The name of your medicine is ZUVOG. We refer to them as ZUVOG TABLETS or ZUVOG throughout this leaflet.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any more questions, please ask your doctor or your pharmacist. '
- This medicine has been prescribed for you personally and you should not pass it on to anyone else. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects get serious, or if you notice any side effects that are not listed in the leaflet, please tell your doctor or pharmacist.
In this leaflet:
1. What ZUVOG Tablets are and what they are used for
2. What you need to know before you take ZUVOG Tablets
3. How to take ZUVOG Tablets
4. Possible side effects
5. How to store ZUVOG Tablets
6. Contents of the pack and other information
1. What ZUVOG Tablets are and What they are used for
Zuvog contains the active substance Voglibose, which is used to control postprandial (after-meal) blood sugar levels in patients with type 2 diabetes mellitus. It is prescribed when diet, exercise, or other medications do not adequately control blood sugar levels.
Voglibose works by blocking certain enzymes in your small intestine that break down complex sugars into simple sugars like glucose. By doing this, it slows down the absorption of sugar into your bloodstream after meals, helping to control blood sugar levels.
2. What You Need to Know Before You Take Zuvog Tablets
Do not take Zuvog if you:
- Are allergic to Voglibose or any of the other ingredients of this medicine.
- Have inflammatory bowel disease, gastrointestinal obstruction, or conditions that may deteriorate due to increased gas formation.
- Have severe ketosis, diabetic coma or pre-coma, severe infection, or hepatic impairment.
Taking other medicines and ZUVOG
- Antidiabetic Medications: Co-administration with sulfonylureas or insulin can enhance the hypoglycemic effect, increasing the risk of hypoglycemia. Close monitoring of blood glucose levels is recommended.
- Beta Blockers: These medications can mask the symptoms of hypoglycemia, making it harder to recognize low blood sugar levels.
- Keratolytics and MAOIs: These can enhance the hypoglycemic effect of Voglibose.
- Anti-lipidemic Agents: These can enhance the hypoglycemic effect of Voglibose.
- Sympathomimetics or Adrenergic Agonists: These can reduce the hypoglycemic effect of Voglibose.
- Adrenocorticotropic and Thyroid Hormones: These can reduce the hypoglycemic effect of Voglibose.
Pregnancy
Voglibose is generally not recommended during pregnancy due to insufficient well-controlled studies in pregnant women. It should only be used if specifically advised by a healthcare professional in rare, carefully evaluated situations.
Breast-feeding
The use of Voglibose during breastfeeding is not well-studied, and there is limited data available on its safety for nursing mothers and infants. It is generally recommended to avoid Voglibose during breastfeeding unless absolutely necessary.
Driving and using machines
Voglibose does not usually affect your ability to drive or use machines. However, if you experience symptoms of hypoglycemia, such as dizziness or confusion, avoid these activities until you feel better.
3. How to Take Efnocar Tablets
Dosage:
The usual dose for adults: One tablet of 0.2 mg three times daily before meals. In some cases,
the dose may be increased to 0.3 mg three times daily.
For type 1 diabetes, Voglibose is administered with insulin.
Method of administration:
Take Zuvog immediately before meals to slow the absorption of carbohydrates and prevent sudden spikes in blood sugar.
If you take more ZUVOG Tablets than you should
Taking more Zuvog Tablets than prescribed can lead to an overdose. While Voglibose is unlikely to cause hypoglycemia (low blood sugar) in overdose, it can cause significant gastrointestinal discomfort. Abdominal discomfort, diarrhea, Flatulence and Bloating may occur. Do not panic. Stay calm and seek medical advice immediately. Contact your doctor or go to the nearest hospital emergency department. Take the medicine packaging with you to show the healthcare professionals. Treatment is generally supportive and symptomatic, focusing on relieving gastrointestinal symptoms.
If you forget to take Zuvog Tablets
If you forget to take a tablet, take one as soon as you remember, unless it is nearly time to take the next one. Never take two doses together. Take the remaining doses at the correct time.
If you stop taking Zuvog Tablets
Take this medicine for as long as your doctor tells you to, as you may become unwell if you stop.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Common side effects: Gastrointestinal issues such as flatulence, abdominal distension, diarrhea, and abdominal pain.
Serious side effects:
Hypoglycemia, especially when taken with other anti-diabetic medications.
Hepatotoxicity and skin reactions.
Tell your doctor or pharmacist if you notice any other effects not listed.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top t end of the home page.
Website link: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to Store Efnocar Tablets
Do not use the tablets after the end of the expiry month (use-by date) shown on the product packaging.
Store below 25°C. Protected from moisture.
Keep this medicine out of the sight and reach of children
6. Contents of the Pack and Other Information
What ZUVOG Tablets contain
The active substance is Voglibose
Each tablet contains 0.2/0.3 mg of Voglibose.
Packaging: 10 Blister strips of 10 tablets each
For More Information About This Product
Zuvog 0.2 mg Tablet
1.0 Generic Name
Voglibose Tablets 0.2mg / 0.3mg
2.0 Qualitative and quantitative composition
Each uncoated tablet contains:
Voglibose……………….0.2mg / 0.3mg
Excipients q.s.
3.0 Dosage form and strength
Dosage Form: Tablets.
Dosage Strength: Voglibose 0.2 mg and 0.3 mg per tablet.
4.0 Clinical particulars
4.1 Therapeutic Indication
For improvement of post prandial hyperglycaemia in diabetic mellitus only when diet and/or exercise or oral hypoglycemic drugs or insulin preparations in addition to diet and /or exercise do not result adequate glycemic control.
4.2 Posology and method of administration
ZUVOG is orally administered in a single dose of 0.2 mg three times daily just before or with each meal.
ZUVOG should be administered in conjunction with diet treatment or diet plus a sulphonylurea.
In case of inadequate effect, the single dose may be increased up to 0.3 mg under close observation of the course of the disease.
4.3 Contraindication
- Hypersensitivity to voglibose or to any component of the formulation.
- Diabetic ketoacidosis, diabetic pre-coma.
- Severe infection, before and after surgery, serious trauma.
- Gastrointestinal obstruction or predisposed to it
4.4 Special warnings and precautions for use
Hypoglycaemia
While on voglibose tablets, patients should be instructed and explained to recognize hypoglycaemic symptoms and its management.
Loss of Control of Blood Glucose
When diabetes patients are exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of control of blood glucose may occur. At such times, temporary insulin therapy may be necessary.
4.5 Drugs interactions
Anti-Diabetic Drugs: When voglibose is used in combination with derivative(s) of sulfonylamide, sulfonylurea or biguanide, or with insulin, hypoglycemic symptoms may occur. Therefore, when used in combination with any of these drugs, care should be taken, such as to initiate therapy with lower dosage.
Drugs Affecting Glycemic Control: When voglibose is administered concomitantly with drugs that enhance or diminish the hypoglycemic action of antidiabetic drugs, caution should be taken as this might additionally delay the action of voglibose on the absorption of carbohydrates. Examples of drugs enhancing the hypoglycemic action of antidiabetic drugs include alpha-blockers, salicylic acid preparations, monoamine oxidase inhibitors, and fibrate derivatives. Examples of drugs diminishing the hypoglycemic action of antidiabetic drugs include epinephrine, adrenocortical hormone, and thyroid hormone.
Warfarin: Voglibose does not affect the pharmacokinetics of warfarin; hence, it can be safely administered along with warfarin.
4.6 Use in special populations
Pregnant Women
Pregnancy Category B. The safety of voglibose in pregnancy has not been established. Animal studies do not indicate harmful effects with respect to pregnancy, embryonal or fetal development, parturition or post-natal development. However, no adequate and well controlled studies have been done in pregnant women. Therefore, voglibose should be given to pregnant women only when the potential benefits to the mother outweigh the possible hazards to the fetus.
Lactating Women
Animal studies (e.g., rats) have revealed a suppressive action of voglibose on body weight increase in newborns probably due to suppression of milk production due to reduced carbohydrate absorption. Although the levels of voglibose reached in human milk are exceedingly low, it is recommended that voglibose may not be administered to lactating women. When the administration of voglibose is unavoidable, nursing should be discontinued.
Paediatric Patients
The safety and effectiveness of voglibose in children has not been established. Thus, Zuvog Tablets are not recommended for use in paediatric patients.
Geriatric
Patients Since elderly patients generally have a physiological hypofunction, it is desirable to initiate therapy with lower dosage. Moreover, this drug should be carefully administered under close observation, throughout the course of the disease, with careful attention to the blood sugar level and the onset of gastrointestinal symptoms.
Renal Impairment Patients
Voglibose is poorly absorbed after oral doses and renal excretion is negligible. Thus, in general, no dosage adjustment is required in patients with renal dysfunction.
Effects on ability to drive and use machines
4.7 Effects on ability to drive and use machines
With voglibose, effect on ability to drive a vehicle or operating machinery has not been reported.
4.8 Undesirable effects
- Gastrointestinal: Gastrointestinal adverse events such as diarrhoea, loose stools, abdominal pain, constipation, anorexia, nausea, vomiting, or heartburn may occur with the use of voglibose. Also, abdominal distention, increased flatus, and intestinal obstruction like symptoms due to an increase in intestinal gas may occur with use of voglibose.
- Hypersensitivity: Rash and pruritus may rarely occur. In such cases, voglibose should be discontinued immediately.
- Hepatic: When voglibose is administered to patients with liver cirrhosis, hyperammonia may worsen with the development of constipation followed by disturbance of consciousness.
- Laboratory Tests: Elevation of SGOT (serum glutamate oxaloacetate), SGPT (serum glutamate pyruvate transaminase), LDH (lactate dehydrogenase), alpha-GPT (alpha-glutamate pyruvate transaminase) or alkaline phosphatase may infrequently occur.
- Hypoglycemia: When voglibose is used in combination with other antidiabetic drugs, hypoglycemia may occur (0.1% to <5%).
- Psychoneurologic: Headache may rarely occur.
- Hematologic: Anemia, thrombocytopenia, and leucopoenia may rarely occur.
- Others: Numbness, edema of face, blurred vision, hot flushes, malaise, weakness, hyperkalemia, increased serum amylase, decreased HDL-cholesterol, diaphoresis, or alopecia may occur rarely with the use of voglibose.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to:medico@zuventus.com
Website: https://www.zuventus.co.in/drug-safety-reporting
4.9 Overdose
Voglibose is unlikely to produce hypoglycemia in overdose, but abdominal discomfort and diarrhoea may occur. If overdose occurs, supportive and symptomatic treatment should be provided.
5.0 Pharmacological properties
5.1 Mechanism of Action
Voglibose competitively and reversibly inhibits the alpha glucosidase enzymes (e.g., glucoamylase, sucrase, maltase, and isomaltase) in the brush border of the small intestine. Alpha-glucosidase enzymes are essential for hydrolysis/decomposition of complex carbohydrates (starch, dextrin, polysaccharides, and disaccharides) into simpler carbohydrates (such as glucose/dextrose or fructose). Inhibition of these enzymes leads to delay in the absorption of glucose into the bloodstream resulting in improvement of post-prandial hyperglycemia.
5.2 Pharmacodynamic properties
Voglibose exerts its activity in the intestinal tract. Alpha glucosidase enzyme normally converts complex carbohydrates into simple monosaccharides (glucose) which can be absorbed through the intestine.
The action of voglibose depends on an inhibition of intestinal enzymes (alpha-glucosidase) involved in the degradation of ingested disaccharides, oligosaccharides, and polysaccharides into monosaccharides. Inhibition of these enzyme systems reduces/delays the rate of digestion of complex carbohydrates. As there is delay in digestion of complex carbohydrates, monosaccharides release slowly and hence absorbed more slowly into the blood i.e., less glucose absorption from intestine into the blood circulation. Voglibose, thus dose dependently reduces the postprandial rise in blood glucose level.
Although voglibose reduces the impact of complex carbohydrates on blood sugar level, it does not affect/inhibit absorption of glucose from the intestine.
Alpha-glucosidase inhibitors such as voglibose do not stimulate insulin release and therefore do not result in hypoglycemia. Voglibose improves post-prandial hyperglycemia and thereby lowers abnormally high levels of glycosylated hemoglobin. Voglibose is highly useful in elderly patients or in patients with predominantly post-prandial hyperglycemia.
5.3 Pharmacokinetic properties
Absorption: Voglibose is poorly absorbed after oral doses. Plasma concentrations after oral doses have usually been undetectable. Following repeated administration to healthy subjects (n=6) in a single dose of 0.2 mg, 3 times a day, for 7 consecutive days, voglibose was not detected in plasma or urine. Similarly, when voglibose was administered to healthy male adults (n=10) as a single dose of 2 mg, voglibose was not detected in plasma or urine.
Distribution: After ingestion of voglibose, the majority of active unchanged drug remains in the lumen of the gastrointestinal tract to exert its pharmacological activity.
Metabolism: Voglibose is metabolized by intestinal enzymes and by the microbial flora. Excretion: Voglibose is mainly excreted in the feces.
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
A study demonstrated that voglibose can potentiate CCl4 (carbon tetrachloride) and APAP (acetaminophen) hepatotoxicity in rats by inducing hepatic CYP2E1.
7.0 Description
Voglibose is an alpha-glucosidase inhibitor used to manage postprandial blood glucose levels in patients with type 2 diabetes mellitus.
Chemical name: (1S, 2S, 3R, 4S, 5S)-5-(1,3-dihydroxypropan-2-ylamino)-1-(hydroxymethyl) cyclohexane-1,2,3,4-tetrol.
Molecular formula: C10H21NO7
Molecular mass: 267.28 g/mol.
Structural formula:
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
Zuvog 0.2 / 0. 3: A blister strip of 10 tablets
8.4 Storage and handing instructions
Store below 25°C. Protected from moisture.
9.0 Patient Counselling Information
- Take this medicine exactly as prescribed by your doctor. Do not change the dose or stop therapy without consulting your doctor.
- Pregnant women and breastfeeding mothers should not use this medicine without doctor consultation.
- Don’t medicine for the treatment of diabetic ketoacidosis or diabetic pre-coma.
- This medicine is not advisable for use in children.
- Avoid use of this medicine during severe infection or before and after surgery or in serious trauma cases.
12.0 Date of revision
10/10/2024
The name of your medicine is ZUVOG. We refer to them as ZUVOG TABLETS or ZUVOG throughout this leaflet.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any more questions, please ask your doctor or your pharmacist. '
- This medicine has been prescribed for you personally and you should not pass it on to anyone else. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects get serious, or if you notice any side effects that are not listed in the leaflet, please tell your doctor or pharmacist.
In this leaflet:
1. What ZUVOG Tablets are and what they are used for
2. What you need to know before you take ZUVOG Tablets
3. How to take ZUVOG Tablets
4. Possible side effects
5. How to store ZUVOG Tablets
6. Contents of the pack and other information
1. What ZUVOG Tablets are and What they are used for
Zuvog contains the active substance Voglibose, which is used to control postprandial (after-meal) blood sugar levels in patients with type 2 diabetes mellitus. It is prescribed when diet, exercise, or other medications do not adequately control blood sugar levels.
Voglibose works by blocking certain enzymes in your small intestine that break down complex sugars into simple sugars like glucose. By doing this, it slows down the absorption of sugar into your bloodstream after meals, helping to control blood sugar levels.
2. What You Need to Know Before You Take Zuvog Tablets
Do not take Zuvog if you:
- Are allergic to Voglibose or any of the other ingredients of this medicine.
- Have inflammatory bowel disease, gastrointestinal obstruction, or conditions that may deteriorate due to increased gas formation.
- Have severe ketosis, diabetic coma or pre-coma, severe infection, or hepatic impairment.
Taking other medicines and ZUVOG
- Antidiabetic Medications: Co-administration with sulfonylureas or insulin can enhance the hypoglycemic effect, increasing the risk of hypoglycemia. Close monitoring of blood glucose levels is recommended.
- Beta Blockers: These medications can mask the symptoms of hypoglycemia, making it harder to recognize low blood sugar levels.
- Keratolytics and MAOIs: These can enhance the hypoglycemic effect of Voglibose.
- Anti-lipidemic Agents: These can enhance the hypoglycemic effect of Voglibose.
- Sympathomimetics or Adrenergic Agonists: These can reduce the hypoglycemic effect of Voglibose.
- Adrenocorticotropic and Thyroid Hormones: These can reduce the hypoglycemic effect of Voglibose.
Pregnancy
Voglibose is generally not recommended during pregnancy due to insufficient well-controlled studies in pregnant women. It should only be used if specifically advised by a healthcare professional in rare, carefully evaluated situations.
Breast-feeding
The use of Voglibose during breastfeeding is not well-studied, and there is limited data available on its safety for nursing mothers and infants. It is generally recommended to avoid Voglibose during breastfeeding unless absolutely necessary.
Driving and using machines
Voglibose does not usually affect your ability to drive or use machines. However, if you experience symptoms of hypoglycemia, such as dizziness or confusion, avoid these activities until you feel better.
3. How to Take Efnocar Tablets
Dosage:
The usual dose for adults: One tablet of 0.2 mg three times daily before meals. In some cases,
the dose may be increased to 0.3 mg three times daily.
For type 1 diabetes, Voglibose is administered with insulin.
Method of administration:
Take Zuvog immediately before meals to slow the absorption of carbohydrates and prevent sudden spikes in blood sugar.
If you take more ZUVOG Tablets than you should
Taking more Zuvog Tablets than prescribed can lead to an overdose. While Voglibose is unlikely to cause hypoglycemia (low blood sugar) in overdose, it can cause significant gastrointestinal discomfort. Abdominal discomfort, diarrhea, Flatulence and Bloating may occur. Do not panic. Stay calm and seek medical advice immediately. Contact your doctor or go to the nearest hospital emergency department. Take the medicine packaging with you to show the healthcare professionals. Treatment is generally supportive and symptomatic, focusing on relieving gastrointestinal symptoms.
If you forget to take Zuvog Tablets
If you forget to take a tablet, take one as soon as you remember, unless it is nearly time to take the next one. Never take two doses together. Take the remaining doses at the correct time.
If you stop taking Zuvog Tablets
Take this medicine for as long as your doctor tells you to, as you may become unwell if you stop.
4. Possible Side Effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Common side effects: Gastrointestinal issues such as flatulence, abdominal distension, diarrhea, and abdominal pain.
Serious side effects:
Hypoglycemia, especially when taken with other anti-diabetic medications.
Hepatotoxicity and skin reactions.
Tell your doctor or pharmacist if you notice any other effects not listed.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top t end of the home page.
Website link: https://www.zuventus.co.in/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to Store Efnocar Tablets
Do not use the tablets after the end of the expiry month (use-by date) shown on the product packaging.
Store below 25°C. Protected from moisture.
Keep this medicine out of the sight and reach of children
6. Contents of the Pack and Other Information
What ZUVOG Tablets contain
The active substance is Voglibose
Each tablet contains 0.2/0.3 mg of Voglibose.
Packaging: 10 Blister strips of 10 tablets each