Elriz Syrup


1.0 Generic name
Levocetirizine syrup
2.0 Qualitative and quantitative composition
Each 5 ml syrup contains:
Levocetirizine Hydrochloride 2.5 mg
Excipients …………………. q.s.
In a flavored syrup base.
3.0 Dosage form and strength
Oral syrup, Levocetirizine 2.5mg/5ml
4.0 Clinical particulars
4.1 Therapeutic Indication
For allergic rhinitis and chronic urticaria.
4.2 Posology and method of administration
Adults and adolescents 12 years and above:
The daily recommended dose is 5 mg (10 ml of solution).
Elderly
Adjustment of the dose is recommended in elderly patients with moderate to severe renal impairment.
Renal impairment
The dosing intervals must be individualised according to renal function (eGFR – estimated Glomerular Filtration Rate). Refer to the following table and adjust the dose as indicated.
Dosing adjustments for patients with impaired renal function:
| Group | eGFR (ml/min) | Dosage and frequency |
| Normal renal function | ≥ 90 | 5 mg once daily |
| Mildly decreased renal function/td> | 60 – < 90 | 5 mg once daily |
| Moderately decreased renal function | 30 – < 60 | 5 mg once every 2 days |
| Severely decreased renal function | 15 – < 30 (not requiring dialysis) | 5 mg once every 3 days |
| End stage renal disease (ESRD) | < 15 (requiring dialysis treatment) | Contra-indicated |
In paediatric patients suffering from renal impairment, the dose will have to be adjusted on an individual basis taking into account the renal clearance of the patient and his body weight. There are no specific data for children with renal impairment.
Hepatic impairment
No dose adjustment is needed in patients with solely hepatic impairment. In patients with hepatic impairment and renal impairment, adjustment of the dose is recommended (see Renal impairment above).
Paediatric population
Children aged 6 to 12 years:
The daily recommended dose is 5 mg (10 ml of solution).
Children aged 2 to 6 years:
The daily recommended dose is 2.5 mg to be administered in 2 intakes of 1.25 mg (2.5 ml of solution twice daily).
Even if some clinical data are available in children aged 6 months to 12 years, these data are not sufficient to support the administration of levocetirizine to infants and toddlers aged less than 2 years.
Method of administration
An oral syringe is included in the package. The appropriate volume of oral solution should be measured with the oral syringe, and poured in a spoon or in a glass of water. The oral solution must be taken orally immediately after dilution, and may be taken with or without food.
Duration of use:
Intermittent allergic rhinitis (symptoms experienced for less than four days a week or for less than four weeks a year) has to be treated according to the disease and its history; it can be stopped once the symptoms have disappeared and can be restarted again when symptoms reappear. In case of persistent allergic rhinitis (symptoms experienced for more than four days a week or for more than four weeks a year), continuous therapy can be proposed to the patient during the period of exposure to allergens.
There is clinical experience with the use of levocetirizine for treatment periods of at least 6 months. In chronic urticaria and chronic allergic rhinitis, there is clinical experience of use of cetirizine (racemate) for up to one year.
4.3 Contraindications
Hypersensitivity to the active substance, to cetirizine, to hydroxyzine, to any other piperazine derivatives or to any of the other excipients.
Patients with end stage renal disease with estimated Glomerular Filtration Rate (eGFR) below 10 ml/min (requiring dialysis treatment).
4.4 Special warnings and precautions for use
Precaution is recommended with concurrent intake of alcohol.
Elriz contains methyl parahydroxybenzoate and propyl parahydroxybenzoate which may cause allergic reactions (possibly delayed).
Caution should be taken in patients with epilepsy and patients at risk of convulsion as levocetirizine may cause seizure aggravation.
Elriz contains maltitol liquid
Patients with rare hereditary problems of fructose intolerance should not take this medicine.
Caution should be taken in patients with predisposing factors of urinary retention (e.g. spinal cord lesion, prostatic hyperplasia) as levocetirizine may increase the risk of urinary retention.
Response to allergy skin tests are inhibited by antihistamines and a wash-out period (of 3 days) is required before performing them.
Pruritus may occur when levocetirizine is stopped even if those symptoms were not present before treatment initiation. The symptoms may resolve spontaneously. In some cases, the symptoms may be intense and may require treatment to be restarted. The symptoms should resolve when the treatment is restarted.
Pediatric population
Even if some clinical data are available in children aged 6 months to 12 years, these data are not sufficient to support the administration of levocetirizine to infants and toddlers aged less than 2 years.
4.5 Drugs interactions
No interaction studies have been performed with levocetirizine (including no studies with CYP3A4 inducers); studies with the racemate compound cetirizine demonstrated that there were no clinically relevant adverse interactions (with antipyrine, azithromycin, cimetidine, diazepam, erythromycin, glipizide, ketoconazole and pseudoephedrine). A small decrease in the clearance of cetirizine (16%) was observed in a multiple dose study with theophylline (400 mg once a day); while the disposition of theophylline was not altered by concomitant cetirizine administration.
In a multiple dose study of ritonavir (600 mg twice daily) and cetirizine (10 mg daily), the extent of exposure to cetirizine was increased by about 40% while the disposition of ritonavir was slightly altered (-11%) further to concomitant cetirizine administration.
The extent of absorption of levocetirizine is not reduced with food, although the rate of absorption is decreased.
In sensitive patients, the concurrent administration of cetirizine or levocetirizine and alcohol or other CNS depressants may cause additional reductions in alertness and impairment of performance.
4.6 Use in special populations
Pregnancy
There are no or limited amount of data (less than 300 pregnancy outcomes) from the use of levocetirizine in pregnant women. However, for cetirizine, the racemate of levocetirizine, a large amount of data (more than 1000 pregnancy outcomes) on pregnant women indicate no malformative or feto/neonatal toxicity. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal development, parturition or postnatal development.
The use of levocetirizine may be considered during pregnancy, if necessary.
Breast-feeding
Cetirizine, the racemate of levocetirizine, has been shown to be excreted in human. Therefore, the excretion of levocetirizine in human milk is likely. Adverse reactions associated with levocetirizine may be observed in breastfed infants. Therefore, caution should be exercised when prescribing levocetirizine to lactating women.
Fertility
For levocetirizine no clinical data are available.
4.7 Effects on ability to drive and use machines
Comparative clinical trials have revealed no evidence that levocetirizine at the recommended dose impairs mental alertness, reactivity or the ability to drive and use machines.
Nevertheless, some patients could experience somnolence, fatigue and asthenia under therapy with levocetirizine. Therefore, patients intending to drive, engage in potentially hazardous activities or operate machinery should take their response to the medicinal product into account.
4.8 Undesirable effects
Clinical studies
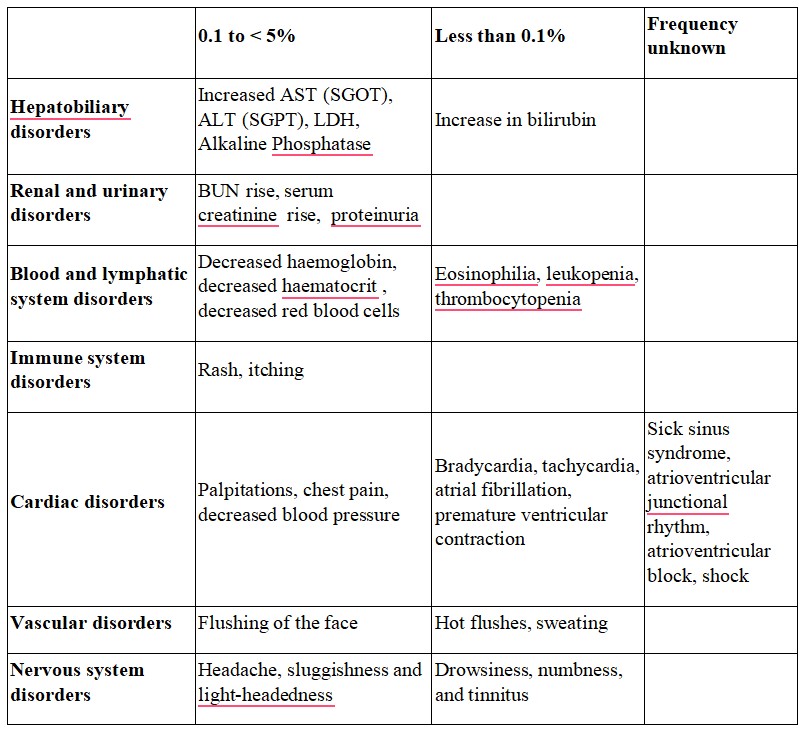
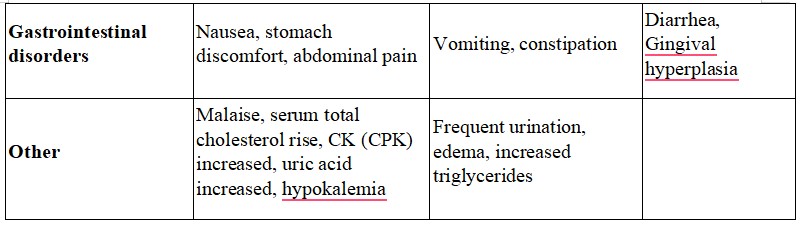
Adults and adolescents above 12 years of age In therapeutic studies in women and men aged 12 to 71 years, 15.1% of the patients in the levocetirizine 5 mg group had at least one adverse drug reaction compared to 11.3% in the placebo group. 91.6 % of these adverse drug reactions were mild to moderate. In therapeutic trials, the dropout rate due to adverse events was 1.0% (9/935) with levocetirizine 5 mg and 1.8% (14/771) with placebo. Clinical therapeutic trials with levocetirizine included 935 subjects exposed to the medicinal product at the recommended dose of 5 mg daily. From this pooling, following incidence of adverse drug reactions were reported at rates of 1% or greater (common: ≥ 1/100 to < 1/10) under levocetirizine 5 mg or placebo:
| Preferred Term (WHOART) | Placebo (n =771) | Levocetirizine 5 mg (n = 935) |
| Headache | 25 (3.2%) | 24 (2.6%) |
| Somnolence | 11 (1.4%) | 49 (5.2%) |
| Mouth dry | 12 (1.6%) | 24 (2.6%) |
| Fatigue | 9 (1.2%) | 23 (2.5%) |
Further uncommon incidences of adverse reactions (uncommon ≥ 1/1000 to < 1/100) like asthenia or abdominal pain were observed.
The incidence of sedating adverse drug reactions such as somnolence, fatigue, and asthenia was altogether more common (8.1%) under levocetirizine 5 mg than under placebo (3.1%).
Paediatric population
In two placebo-controlled studies in paediatric patients aged 6-11 months and aged 1 year to less than 6 years, 159 subjects were exposed to levocetirizine at the dose of 1.25 mg daily for 2 weeks and 1.25 mg twice daily respectively. The following incidence of adverse drug reactions was reported at rates of 1% or greater under levocetirizine or placebo.
| System Organ Class and Preferred Term | Placebo (n=83) | Levocetirizine (n=159) |
| Gastrointestinal disorders | ||
| Diarrhoea | 0 | 3(1.9%) |
| Vomiting | 1(1.2%) | 1(0.6%) |
| Constipation | 0 | 2(1.3%) |
| Nervous system disorders | ||
| Somnolence | 2(2.4%) | 3(1.9%) |
| Psychiatric disorders | ||
| Sleep disorder | 0 | 2(1.3%) |
In children aged 6-12 years double blind placebo controlled studies were performed where 243 children were exposed to 5 mg levocetirizine daily for variable periods ranging from less than 1 week to 13 weeks. The following incidence of adverse drug reactions was reported at rates of 1% or greater under levocetirizine or placebo.
| Preferred Term | Placebo (n=240) | Levocetirizine 5mg (n=243) |
| Headache | 5(2.1%) | 2(0.8%) |
| Somnolence | 1(0.4%) | 7(2.9%) |
Post-marketing experience
Adverse reactions from post-marketing experience are per System Organ Class and per frequency. The frequency is defined as follows: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data).
- Immune system disorders:Not known: hypersensitivity including anaphylaxis
- Metabolism and nutrition disorders: Not known: increased appetite
- Psychiatric disorders:Not known: aggression, agitation, hallucination, depression, insomnia, suicidal ideation, nightmare
- Nervous system disorders: Not known: convulsion, paraesthesia, dizziness, syncope, tremor, dysgeusia
- Ear and labyrinth disorders: Not known: vertigo
- Eyes disorders: Not known: visual disturbances, blurred vision, oculogyration
- Cardiac disorders: Not known: palpitations, tachycardia
- Respiratory, thoracic and mediastinal disorders:Not known: dyspnoea
- Gastrointestinal disorders: Not known: nausea, vomiting, diarrhoea
- Hepatobiliary disorders: Not known: hepatitis
- Renal and urinary disorders: Not known: dysuria, urinary retention
- Skin and subcutaneous tissue disorders: Not known: angioneurotic oedema, fixed drug eruption, pruritus, rash, urticarial
- Musculoskeletal, connective tissues, and bone disorders: Not known: myalgia, arthralgia
- General disorders and administration site conditions: Not known: oedema
- Investigations: Not known: weight increased, abnormal liver function tests
- Description of selected adverse reactions After levocetirizine discontinuation, pruritus has been reported.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
4.9 Overdose
Symptoms
Symptoms of overdose may include drowsiness in adults. In children, agitation and restlessness may initially occur, followed by drowsiness.
Management of overdoses
There is no known specific antidote to levocetirizine.
Should overdose occur, symptomatic or supportive treatment is recommended. Gastric lavage may be considered shortly after ingestion of the drug. Levocetirizine is not effectively removed by haemodialysis.
5.0 Pharmacological properties
5.1 Mechanism of Action
Levocetirizine hydrochloride, the active enantiomer of cetirizine, is a second-generation anti-histamine and a potent and selective antagonist of peripheral H1-receptors.
5.2 Pharmacodynamic properties
Pharmacodynamic effects
The pharmacodynamic activity of levocetirizine has been studied in randomised, controlled trials:
In a study comparing the effects of levocetirizine 5 mg, desloratadine 5 mg, and placebo on histamine-induced wheal and flare, levocetirizine treatment resulted in significantly decreased wheal and flare formation which was highest in the first 12 hours and lasted for 24 hours, (p<0.001) compared with placebo and desloratadine.
The onset of action of levocetirizine 5 mg in controlling pollen-induced symptoms has been observed at 1 hour post drug intake in placebo controlled trials in the model of the allergen challenge chamber.
In vitro studies (Boyden chambers and cell layers techniques) show that levocetirizine inhibits eotaxin-induced eosinophil transendothelial migration through both dermal and lung cells. A pharmacodynamic experimental study in vivo (skin chamber technique) showed three main inhibitory effects of levocetirizine 5 mg in the first 6 hours of pollen-induced reaction, compared with placebo in 14 adult patients: inhibition of VCAM-1 release, modulation of vascular permeability and a decrease in eosinophil recruitment.
Clinical efficacy and safety
The efficacy and safety of levocetirizine has been demonstrated in several double-blind, placebo controlled, clinical trials performed in adult patients suffering from seasonal allergic rhinitis, perennial allergic rhinitis, or persistent allergic rhinitis. Levocetirizine has been shown to significantly improve symptoms of allergic rhinitis, including nasal obstruction in some studies.
A 6-month clinical study in 551 adult patients (including 276 levocetirizine-treated patients) suffering from persistent allergic rhinitis (symptoms present 4 days a week for at least 4 consecutive weeks) and sensitized to house dust mites and grass pollen demonstrated that levocetirizine 5 mg was clinically and statistically significantly more potent than placebo on the relief from the total symptom score of allergic rhinitis throughout the whole duration of the study, without any tachyphylaxis. During the whole duration of the study, levocetirizine significantly improved the quality of life of the patients.
In a placebo-controlled clinical trial including 166 patients suffering from chronic idiopathic urticaria, 85 patients were treated with placebo and 81 patients with levocetirizine 5 mg once daily over six weeks. Treatment with levocetirizine resulted in significant decrease in pruritus severity over the first week and over the total treatment period as compared to placebo. Levocetirizine also resulted in a larger improvement of health-related quality of life as assessed by the Dermatology Life Quality Index as compared to placebo. Chronic idiopathic urticaria was studied as a model for urticarial conditions. Since histamine release is a causal factor in urticarial diseases, levocetirizine is expected to be effective in providing symptomatic relief for other urticarial conditions, in addition to chronic idiopathic urticaria.
ECGs did not show relevant effects of levocetirizine on QT interval.
5.3 Pharmacokinetic properties
The pharmacokinetics of levocetirizine are linear with dose- and time-independent with low inter-subject variability. The pharmacokinetic profile is the same when given as the single enantiomer or when given as cetirizine. No chiral inversion occurs during the process of absorption and elimination.
Absorption
Levocetirizine is rapidly and extensively absorbed following oral administration. In adults, peak plasma concentrations are achieved 0.9 h after dosing. Steady state is achieved after two days. Peak concentrations are typically 270 ng/ml and 308 ng/ml following a single and a repeated 5 mg o.d. dose, respectively. The extent of absorption is dose-independent and is not altered by food, but the peak concentration is reduced and delayed.
Distribution
No tissue distribution data are available in humans, neither concerning the passage of levocetirizine through the blood-brain-barrier.
In rats and dogs, the highest tissue levels are found in liver and kidneys, the lowest in the CNS compartment. In humans, levocetirizine is 90% bound to plasma proteins. The distribution of levocetirizine is restrictive, as the volume of distribution is 0.4 l/kg.
Biotransformation
The extent of metabolism of levocetirizine in humans is less than 14% of the dose and therefore differences resulting from genetic polymorphism or concomitant intake of enzyme inhibitors are expected to be negligible. Metabolic pathways include aromatic oxidation, N- and Odealkylation and taurine conjugation. Dealkylation pathways are primarily mediated by CYP 3A4 while aromatic oxidation involved multiple and/or unidentified CYP isoforms. Levocetirizine had no effect on the activities of CYP isoenzymes 1A2, 2C9, 2C19, 2D6, 2E1 and 3A4 at concentrations well above peak concentrations achieved following a 5 mg oral dose. Due to its low metabolism and absence of metabolic inhibition potential, the interaction of levocetirizine with other substances, or vice-versa, is unlikely.
Elimination
The plasma half-life in adults is 7.9 ± 1.9 hours. The half-life is shorter in small children.
The mean apparent total body clearance in adults is 0.63 ml/min/kg. The major route of excretion of levocetirizine and metabolites is via urine, accounting for a mean of 85.4% of the dose. Excretion via faeces accounts for only 12.9% of the dose. Levocetirizine is excreted both by glomerular filtration and active tubular secretion.
6.0 Nonclinical Properties
6.1 Animal Toxicology or Pharmacology
No carcinogenicity studies have been performed with levocetirizine. However, evaluation of cetirizine carcinogenicity studies are relevant for determination of the carcinogenic potential of levocetirizine. In a 2-year carcinogenicity study, in rats, cetirizine was not carcinogenic at dietary doses up to 20 mg/kg (approximately 15 times the maximum recommended daily oral dose in adults, approximately 10 times the maximum recommended daily oral dose in children 6 to 11 years of age and approximately 15 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). In a 2-year carcinogenicity study in mice, cetirizine caused an increased incidence of benign hepatic tumors in males at a dietary dose of 16 mg/kg (approximately 6 times the maximum recommended daily oral dose in adults, approximately 4 times the maximum recommended daily oral dose in children 6 to 11 years of age, and approximately 6 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m basis). No increased incidence of benign tumors was observed at a dietary dose of 4 mg/kg (approximately 2 times the maximum recommended daily oral dose in adults, equivalent to the maximum recommended daily oral dose in children 6 to 11 years of age and approximately 2 times the maximum recommended daily oral dose in children 6 months to 5 years of age on a mg/m2 basis). The clinical significance of these findings during long-term use of levocetirizine dihydrochloride is not known.
Levocetirizine was not mutagenic in the Ames test, and not clastogenic in the human lymphocyte assay, the mouse lymphoma assay, and in vivo micronucleus test in mice. In a fertility and general reproductive performance study in mice, cetirizine did not impair fertility at an oral dose of 64 mg/kg (approximately 25 times the recommended daily oral dose in adults on a mg/m² basis)
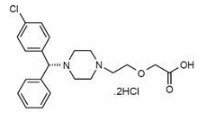
7.0 Description
Levocetirizine dihydrochloride, the active component of Elriz oral solution, is an orally active H1‑receptor antagonist.
The chemical name is (R)-[2-[4-[(4-chlorophenyl) phenylmethyl]-1-piperazinyl] ethoxy] acetic acid dihydrochloride. Levocetirizine dihydrochloride is the R enantiomer of cetirizine hydrochloride, a racemic compound with antihistaminic properties.
The empirical formula of levocetirizine dihydrochloride is C21H25ClN2O3•2HCl.
The molecular weight is 461.82
Chemical structure is shown below:
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
A bottle of 30 ml
8.4 Storage and handing instructions
Store below 25°C. Protect from light.
9.0 Patient Counselling Information
Somnolence
Caution patients against engaging in hazardous occupations requiring complete mental alertness, and motor coordination such as operating machinery or driving a motor vehicle after ingestion of levocetirizine dihydrochloride.
Concomitant
Use of Alcohol and other Central Nervous System Depressants Instruct patients to avoid concurrent use of levocetirizine hydrochloride with alcohol or other central nervous system depressants because additional reduction in mental alertness may occur.
Dosing of Levocetirizine hydrochloride syrup
Do not exceed the recommended daily dose. Advise patients to not ingest more than the recommended dose of levocetirizine hydrochloride because of the increased risk of somnolence at higher doses.
Do not take this medicine, if you are allergic to levocetirizine
Signs of an allergic reaction include a rash, itching or shortness of breath.
Advice patients to talk to your doctor or pharmacist, if you get any side effects. This includes any possible side effects not listed in this leaflet.
Advice patients if they have any further questions, ask doctor or pharmacist.
12.0 Date of revision
30.08.2024
About leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What Elriz is and what it is used for
- What you need to know before you take Elriz
- How to take Elriz
- Possible side effects
- How to store Elriz
- Contents of the pack and other information
1. What Elriz is and what it is used for
Levocetirizine dihydrochloride is the active ingredient of Elriz. Elriz is an anti-allergic medication. It is used for the treatment of signs of illness (symptoms) associated with:
- allergic rhinitis (including persistent allergic rhinitis)
- nettle rash (urticaria).
2. What you need to know before you take Elriz
Do not take Elriz
- If you are allergic to levocetirizine dihydrochloride, to cetirizine, to hydroxyzine or any of the other ingredients of this medicine.
- If you have a severe kidney disease requiring dialysis.
Warnings and precautions
Talk to your doctor or pharmacist before taking Elriz.
If you are likely to be unable to empty your bladder (with conditions such as spinal cord injury or enlarged prostate), please ask your doctor for advice.
If you suffer from epilepsy or are at risk of convulsions, please ask your doctor for advice as use of Elriz may cause seizure aggravation.
If you are scheduled for allergy testing, ask your doctor if you should stop taking Elriz for several days before testing. This medicine may affect your allergy test results.
Children
The use of Elriz is not recommended for infants and children under 2 years of age.
Other medicines and Elriz
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
Elriz with food, drink and alcohol
Caution is advised if Elriz is taken at the same time as alcohol or other agents acting on the brain. In sensitive patients, the concurrent administration of Elriz and alcohol or other agents acting on the brain may cause additional reductions in alertness and impairment of performance.
Elriz can be taken with or without food.
Pregnancy, breast-feeding and fertility
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Driving and using machines
Some patients being treated with Elriz may experience somnolence/drowsiness, tiredness and exhaustion. Use caution when driving or operating machinery until you know how this medicine affects you. However, special tests have revealed no impairment of mental alertness, the ability to react or the ability to drive in healthy test persons after taking levocetirizine in the recommended dosage.
3. How to take Elriz
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
The recommended dose is:
Adults and adolescents from the age of 12 years: 10 ml solution once daily.
Special dosage instructions for specific populations:
Renal and hepatic impairment
Patients with impaired kidney function may be given a lower dose according to the severity of their kidney disease, and in children the dose will also be chosen on the basis of body weight; the dose will be determined by your doctor.
Patients who have a severe kidney disease requiring dialysis must not take Elriz.
Patients who only have impaired liver function should take the usual prescribed dose.
Patients who have both impaired liver and kidney function may be given a lower dose depending on the severity of the kidney disease, and in children the dose will also be chosen on the basis of body weight; the dose will be determined by your doctor.
Elderly patients aged 65 years and above
No adaptation of the dose is necessary in elderly patients, provided their renal function is normal.
Use in children
Children from the age of 6 to 12 years: 10 ml solution once daily.
Children from the age of 2 to 6 years: 2.5 ml solution twice daily.
The administration of Elriz to infants and toddlers aged less than 2 years is not recommended.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Common: may affect up to 1 in 10 people
Dry mouth, headache, tiredness and somnolence/drowsiness
Uncommon: may affect up to 1 in 100 people
Exhaustion and abdominal pain
Not known: frequency cannot be estimated from the available data
Other side effects such as palpitations, increased heart rate, fits, pins and needles, dizziness, syncope, tremor, dysgeusia (distortion of the sense of taste), sensation of rotation or movement, visual disturbances, blurred vision, oculogyration (eyes having uncontrolled circular movements), painful or difficult urination, inability to completely empty the bladder, oedema, pruritus (itchiness), rash, urticaria (swelling, redness and itchiness of the skin), skin eruption, shortness of breath, weight increase, muscular pain, joint pain, aggressive or agitated behaviour, hallucination, depression, insomnia, recurring thoughts of or preoccupation with suicide, nightmare, hepatitis, abnormal liver function, vomiting, increased appetite, nausea and diarrhea have also been reported. Pruritus (intense itching) upon discontiunation.
At the first signs of a hypersensitivity reaction, stop taking Elriz and tell your doctor. Hypersensitivity reaction symptoms may include swelling of the mouth, tongue, face and/or throat, breathing or swallowing difficulties (chest tightness or wheezing), hives, sudden fall in blood pressure leading to collapse or shock, which may be fatal.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse.This includes any possible side effects not listed in this leaflet. You can also report side effects directly. Website: https://www.zuventus.com/ and click the tab “Drug Safety Reporting” located on the top end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Elriz
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the label and carton after EXP. The expiry date refers to the last day of that month.
This medicinal product does not require any special storage conditions. Do not use 3 months after first opening.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Elriz contains
Composition:
Each 5 ml contains:
Levocetirizine Hydrochloride -2.5 mg
Packing
A bottle of 30 ml