Setolac®-p


1.0 Name of the medicinal product
S (+) Etodolac and Paracetamol Tablets (200mg + 325mg)
2.0 Qualitative and quantitative composition
Each uncoated tablet contains:
S (+) Etodolac 200 mg
Paracetamol IP 325 mg
Excipients q.s
3.0 Dosage form and strength
Tablet
200 mg of S (+) Etodolac and 325 mg of Paracetamol
4.0 Clinical Particulars
4.1 Therapeutic indication
For the symptomatic treatment of acute pain and inflammation in patients with osteoarthritis, rheumatoid arthritis and ankylosing spondylitis.
4.2 Posology and method of administration
Adults: One tablet two times daily orally.
Paediatric population: Not recommended.
Method of administration: For oral administration.
To be taken preferably with or after food. Swallow the tablet whole with a glass of water.
4.3 Contraindications
- Patients with known hypersensitivity to S (+) Etodolac and/or Paracetamol.
- Patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs.
- Patients with severe hepatocellular insufficiency
- Setolac®-P Tablets should not be used in patients with active or history of recurrent peptic ulceration or a history of peptic ulcer disease (with two or more distinct episodes of proven ulceration or bleeding).
- During the last trimester of pregnancy.
- Patients with hepatic failure, active liver disease and renal failure.
- Setolac®-P tablet is not recommended for pediatric use.
4.4 Special warnings and precautions for use
Undesirable effects may be minimised by using the lowest effective dose for the shortest duration necessary to control symptoms.
The use of Setolac®-P Tablets with concomitant NSAIDs including cyclooxygenase-2-selective inhibitors should be avoided.
Cardiovascular and Cerebrovascular Effects
COX-2 selective and non-selective NSAIDs have shown an increased risk of serious cardiovascular (CV) thrombotic events, myocardial infarction, and stroke, which can be fatal. Patients with known CV disease or risk factors for CV disease may be at greater risk. To minimize the potential risk for an adverse CV events in patients treated with an NSAID, the lowest effective dose should be used for the shortest duration possible. NSAIDs, including Etodolac, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy. Fluid retention and edema have been observed in some patients taking NSAIDs. Setolac®-P tablets should be used with caution in patients with fluid retention or heart failure.
Pre-existing Asthma
Patients with asthma may have Aspirin-sensitive asthma. Since cross reactivity, including bronchospasm, between Aspirin and other NSAIDs has been reported in such Aspirin-sensitive patients, Setolac®-P tablets should not be administered to patients with this form of Aspirin sensitivity and should be used with caution in patients with pre-existing asthma.
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. In these patients, administration of a NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. No information is available from controlled clinical studies regarding the use of S (+) Etodolac extended-release tablets in patients with advanced renal disease. Therefore, treatment with Setolac®-P tablets is not recommended in these patients with advanced renal disease.
Hepatic Effects
Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs including S (+) Etodolac. Owing to the presence of Paracetamol, Setolac®-P tablets should be used with caution in patients with hepatocellular insufficiency or non-cirrhotic alcoholic liver disease. Impairment of renal or hepatic functions due to other causes may alter drug metabolism; patients receiving concomitant long term therapy, especially the elderly, should be observed for potential side effects and their drug doses adjusted as needed, or the drug discontinued.
Gastrointestinal Effects
NSAIDs, including Etodolac, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. Setolac®-P tablets should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. To minimize the potential risk for an adverse GI event in patients, the lowest effective dose should be used for the shortest possible duration. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population
Hematological Effects
Anemia is sometimes seen in patients receiving NSAIDs, including Etodolac, due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including Etodolac, should have their hemoglobin or haematocrit checked if they exhibit any signs or symptoms of anemia. NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Patients receiving Setolac®-P tablets who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored.
SLE and mixed connective tissue disease:
In patients with systemic lupus erythematous (SLE) and mixed connective tissue disorders there may be an increased risk of aseptic meningitis.
Dermatological:
Serious skin reactions, some of them fatal, including exfoliative dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis, have been reported very rarely in association with the use of NSAIDs (see section 4.8). Patients appear to be at highest risk for these reactions early in the course of therapy: the onset of the reaction occurring in the majority of cases within the first month of treatment. Tablets should be discontinued at the first appearance of the skin rash, mucosal lesions, or any other sign of hypersensitivity
Anaphylactoid Reactions
As with other NSAIDs, anaphylactoid reactions may occur in patients without known prior exposure to Etodolac. Setolac®-P tablets should not be given to patients with the Aspirin triad.
Other Precautions
- S (+) Etodolac cannot be expected to substitute corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation.
- The pharmacological activity of Setolac®-P in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
- Setolac®-P tablets should be used with caution in patients with severe renal insufficiency (creatinine clearance 30 mL/min), Glucose-6-Phosphate Dehyrogenase (G6PD) deficiency, chronic alcoholism, excessive alcohol intake (3 or more alcoholic drinks every day), anorexia, bulimia or cachexia; chronic malnutrition (low reserves of hepatic glutathione), dehydration, and hypovolemia.
4.5 Drug interactions
ACE inhibitors: Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE inhibitors.
Aspirin: When Etodolac is administered with Aspirin, its protein binding is reduced, although the clearance of free Etodolac is not altered.
Busulfan: Busulfan is eliminated from the body via conjugation with Glutathione. Concomitant use with Paracetamol may result in reduced Busulfan clearance.
Other analgesics including cyclooxygenase-2 selective inhibitor: Avoid concomitant use of two or more NSAIDs (including aspirin) as this may increase the risk of adverse effects (see section 4.4)
Anti-hypertensives: Reduced anti-hypertensive effect
Diuretics: Etodolac can reduce the natriuretic effect of furosemide and thiazides in some patients with possible loss of blood pressure control. Caution should be paid to the concomitant intake of enzyme-inducing agents. Induction of metabolism of Paracetamol from enzyme inducers may result in an increased level of hepatotoxic metabolites.
Cardiac glycosides: NSAIDs may exacerbate cardiac failure, reduce GFR and increase plasma glycoside levels.
Lithium: NSAIDs have produced an elevation of plasma Lithium levels and a reduction in renal Lithium clearance. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and Lithium are administered concurrently, subjects should be observed, carefully for signs of Lithium toxicity.
Cyclosporin, Digoxin, Methotrexate: Etodolac, like other NSAIDs, through effects on renal prostaglandins, may cause changes in the elimination of these drugs leading to elevated serum levels of Cyclosporine, Digoxin, Methotrexate, and increased toxicity. Nephrotoxicity associated with Cyclosporine may also be enhanced. Concomitant diflunisal increases Paracetamol plasma concentrations and this may increase hepatotoxicity.
Anti-coagulants: NSAIDs may enhance the effects of anti-coagulants, such as warfarin (see section 4.4)
Anti-platelet agents and selective serotonin reuptake inhibitors (SSRIs): Increased risk of gastrointestinal bleeding (see section 4.4)
Tacrolimus: Possible increased risk of nephrotoxicity when NSAIDs are given with tacrolimus.
Zidovudine: Increased risk of haematological toxicity when NSAIDs are given with zidovudine. There is an evidence of an increased risk of haemarthroses and haemtoma in HIV (+) haemophiliacs receiving concurrent treatment with zidovudine and ibuprofen.
Bilirubin tests can give a false positive result due to the presence of phenolic metabolites of Setolac®-P Tablets in the urine.
Mifepristone: NSAIDs should not be used for 8-12 days after mifepristone administration as NSAIDs can reduce the effect of mifepristone.
Corticosteroids: increased risk of gastrointestinal ulceration or bleeding (see section 4.4)
Quinolone antibiotics: animal data indicate that NSAIDs can increase the risk of convulsions associated with quinolone antibiotics. Patients taking NSAIDs and quinolones may have an increased risk of developing convulsions.
Phenylbutazone: Phenylbutazone causes increase (by about 80%) in the free fraction of Etodolac though the clinical implication of the same is not known.
Cholestyramine: The speed of absorption of paracetamol is reduced by cholestyramine. Therefore, the cholestyramine should not be taken within one hour if maximal analgesia is required.
Metoclopramide and Domperidone: The absorption of paracetamol is increased by metoclopramide and domperidone. However, concurrent use need not be avoided.
Warfarin: The anticoagulant effect of warfarin and other coumarins may be enhanced by prolonged regular use of paracetamol with increased risk of bleeding; occasional doses have no significant effect.
Chloramphenicol: Increased plasma concentration of chloramphenicol.
Phenytoin: Administered concomitantly may result in decreased Paracetamol effectiveness and an increased risk of hepatotoxicity.
Probenecid: Causes an almost 2-fold reduction in clearance of Paracetamol by inhibiting its conjugation with Glucuronic acid.
Salicylamide: May prolong the amount of time Paracetamol stays in the body.
4.6 Use in special populations
Fertility:
The use of Setolac®-P Tablets may impair female fertility and is not recommended in woman attempting to conceive. In women who have difficulties conceiving or who are undergoing investigation of infertility, withdrawal of Setolac®-P Tablets should be considered.
Pregnancy:
In teratology studies, isolated occurrences of alterations in limb development were found and included polydactyly, oligodactyly, syndactyly, and unossified phalanges in rats and oligodactyly and synostosis of metatarsals in rabbits. Drugs which inhibit prostaglandin biosynthesis may cause dystocia and delayed parturition as evidenced by studies in pregnant animals. Congenital abnormalities have been reported in association with NSAID administration in man; however, these are low in frequency and do not appear to follow any discernible pattern. In view of the known effects of NSAIDs on the foetal cardiovascular system, some inhibitors of prostaglandin biosynthesis have been shown to interfere with the risk of closure of the ductus arteriosus, use in the last trimester of pregnancy is contraindicated. The onset of labour may be delayed and the duration increased with an increased bleeding tendency in both mother and child.
From the 20th week of pregnancy onward, etodolac use may cause oligohydramnios resulting from foetal renal dysfunction. This may occur shortly after treatment initiation and is usually reversible upon discontinuation. In addition, there have been reports of ductus arteriosus constriction following treatment in the second trimester, most of which resolved after treatment cessation. Therefore, during the first and second trimester of pregnancy, etodolac should not be given unless clearly necessary. If etodolac is used by a woman attempting to conceive, or during the first and second trimester of pregnancy, the dose should be kept as low and duration of treatment as short as possible. Antenatal monitoring for oligohydramnios and ductus arteriosus constriction should be considered after exposure to etodolac for several days from gestational week 20 onward. Etodolac should be discontinued if oligohydramnios or ductus arteriosus constriction are found.
During the third trimester of pregnancy, all prostaglandin synthesis inhibitors may expose the foetus to:
- cardiopulmonary toxicity (premature constriction/closure of the ductus arteriosus and pulmonary hypertension);
- renal dysfunction (see above); the mother and the neonate, at the end of pregnancy, to:
- possible prolongation of bleeding time, an anti-aggregating effect which may occur even at very low doses.
- inhibition of uterine contractions resulting in delayed or prolonged labour.
Consequently, etodolac is contraindicated during the third trimester of pregnancy.
Lactation:
It is not known whether Setolac®-P is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from S (+) Etodolac, a decision should be made whether to discontinue nursing or to discontinue the drug taking into account the importance of the drug to the mother
Pediatric use:
Safety and effectiveness in pediatric patients below the age of 18 years have not been established.
Geriatric use:
As with any NSAID, caution should be exercised in treating the elderly (65 years and older) and when increasing the dose. Elderly patients may be more sensitive to the anti-prostaglandin effects of NSAIDs (on the gastrointestinal tract and kidneys) than younger patients. In particular, elderly or debilitated patients who receive NSAID therapy seem to have a lower tolerance for gastrointestinal ulceration or bleeding as compared to other individuals, and most spontaneous reports of fatal GI events are in this population. S (+) Etodolac is eliminated primarily by the kidney. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
4.7 Effects on ability to drive and use machines
Setolac®-P may cause dizziness. So, you should not drive or operate heavy machinery if you feel dizzy or not fully alert.
4.8 Undesirable effects
Most frequently reported adverse reactions (approx. 1 - 10%):
Gastrointestinal experiences including: abdominal pain, constipation, diarrhea, dyspepsia, flatulence, gross bleeding / perforation, heartburn, nausea, GI ulcers (gastric / duodenal), vomiting.
Other events including: abnormal renal function, anemia, dizziness, edema, elevated liver enzymes, headaches, increased bleeding time, pruritis, rashes, tinnitus.
Additional NSAID Adverse Experiences Reported Occasionally with Body as a whole: Allergic reaction, anaphylactic / anaphylactoid reactions (including shock), chills, fever, sepsis.
Digestive system: Anorexia, cholestatic hepatitis / jaundice, dry mouth, duodenitis, esophagitis, gastritis, gastric peptic ulcers, glossitis, hepatic failure, hepatitis, hematemesis, intestinal ulceration, jaundice, liver necrosis, melena, pancreatitis, rectal bleeding, stomatitis. Less frequently, gastritis has been observed. Pancreatitis has been reported very rarely.
Hypersensitivity: Hypersensitivity reactions have been reported following treatment with NSAIDs. These may consist of (a)non-specific allergic reactions and anaphylaxis (b)respiratory tract reactivity comprising asthma, aggravated asthma, bronchospasm or dyspnoea, or (c)assorted skin disorders, including rashes of various types, pruritus, urticaria, purpura, angioedema and more rarely exfoliative and bullous dermatoses (including epidermal necrolysis and erythema multiforme).
Cardiovascular and cerebrovascular:
Congestive heart failure, flushing, palpitations, tachycardia, syncope, vasculitis (including necrotizing and allergic). Oedema, hypertension and cardiac failure have been reported in association with NSAID treatment Clinical trial and epidemiological data suggest that use of some NSAIDs (particularly at high doses and in long term treatment) may be associated with an increased risk of arterial thrombotic events (for example myocardial infarction or stroke).
Hepatic:
Abnormal liver function, hepatitis and jaundice Metabolic and nutritional:
Hyperglycemia in previously controlled diabetic patients.
Neurological and special senses: Visual disturbances, photophobia, blurred vision, optic neuritis, headaches, paraethesia, reports of aseptic meningitis (especially in patients with existing auto-immune disorders, such as systemic lupus erythematous, mixed connective tissue disease), with symptoms such as stiff neck, headache, nausea, vomiting, fever or disorientation (See section 4.4), depression, confusion, hallucinations, tinnitus, vertigo, dizziness, malaise, fatigue and drowsiness. Anxiety, confusion, depression, dream abnormalities, insomnia, nervousness, paresthesia, somnolence, tremors, vertigo.
Blood and lymphatic system:
Agranulocytosis, ecchymosis, eosinophilia, hemolytic anemia, leukopenia, neutropenia, pancytopenia, purpura, thrombocytopenia and aplastic anaemia. There have been reports of blood dyscrasias including methaemoglobenaemia and agranulocytosis, but these were not necessarily causality related to paracetamol.
Respiratory system:
Asthma, dyspnea, pulmonary infiltration with eosinophilia.
Dermatological:
Bullous reactions including Stevens Johnson Syndrome and Toxic Epidermal Necrolysis (very rare). Photosensitivity. Angioedema, cutaneous vasculitis with purpura, erythema multiforme, hyperpigmentation, sweating, urticaria, vesiculobullous rash. Fixed drug eruption (FDE) is associated with Paracetamol use.
Urogenital system:
Dysuria, elevated BUN, oliguria / polyuria, proteinuria, renal failure, renal insufficiency, renal papillary necrosis, serum creatinine increase, urinary frequency, Nephrotoxicity in various forms, including interstitial nephritis, nephrotic syndrome.
Reporting of side effects
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: http://www.zuventus.co.in/safety.aspx By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Etodolac
Symptoms include headache, nausea, vomiting, epigastric pain, gastro-intestinal bleeding, rarely diarrhoea, disorientation, excitation, coma, drowsiness, dizziness, tinnitus, fainting, occasionally convulsions. In cases of significant poisoning acute renal failure and liver damage are possible.
Management: Patients should be treated symptomatically as required. Within one hour of ingestion of a potentially toxic amount, activated charcoal should be considered. Alternatively, in adults, gastric lavage should be considered within one hour of indigestion of a potentially life-threatening overdose. Good urine output should be ensured. Renal and liver function should be closely monitored. Patients should be observed for at least four hours after ingestion of potentially toxic amounts. Frequent or prolonged convulsions should be treated with intravenous diazepam. Other measures may be indicated by the patient's clinical condition. The standard practices of gastric lavage, activated charcoal administration and general supportive therapy should be undertaken.
Paracetamol
Symptoms of paracetamol over dosage in the first 24 hours are pallor, nausea, vomiting, anorexia and abdominal pain. Liver damage may become apparent 12 to 48 hours after ingestion. Abnormalities of glucose metabolism and metabolic acidosis may occur. Hypertension, acute renal failure, respiratory depression and coma may occur, but are rare. Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose. In severe poisoning, hepatic failure may progress to encephalopathy, haemorrhage, hypoglycaemia, cerebral oedema, and death. Acute renal failure with acute tubular necrosis, strongly suggested by loin pain, haematuria and proteinuria, may develop even in the absence of severe liver damage. Cardiac arrhythmias and pancreatitis have been reported.
Management: Immediate treatment is essential in the management of paracetamol overdose. Despite a lack of significant early symptoms, patients should be referred to hospital urgently for immediate medical attention. Symptoms may be limited to nausea or vomiting and may not reflect the severity of overdose or the risk of organ damage.
Treatment with activated charcoal should be considered if the overdose has been taken within 1 hour. Plasma paracetamol concentration should be measured at 4 hours or later after ingestion (earlier concentrations are unreliable). Treatment with N-acetylcysteine may be used up to 24 hours after ingestion of paracetamol however, the maximum protective effect is obtained up to 8 hours post ingestion. If required the patient should be given intravenous-N-acetylcysteine, in line with the established dosage schedule. If vomiting is not a problem, oral methionine may be a suitable alternative for remote areas, outside hospital.
Management of patients who present with serious hepatic dysfunction beyond 24 hours from ingestion should be discussed with the NPIS or a liver unit
5.0 Pharmacological Properties
5.1 Mechanism of Action
S (+) Etodolac is the pharmacologically active component of racemate Etodolac. The anti-inflammatory, analgesic, and antipyretic activities of S (+) Etodolac have been observed to be due to inhibition of cyclooxygenase-2 (COX-2) resulting in inhibition of prostaglandin synthesis.
The precise mechanism of the analgesic and antipyretic properties of Paracetamol has yet to be established. It acts both centrally as well as peripherally. Paracetamol is an antipyretic with good analgesic property.
5.2 Pharmacodynamic properties
Etodolac
Inhibition of prostaglandin synthesis and COX-2 selectivity: All non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to inhibit the formation of prostaglandins. It is this action which is responsible both for their therapeutic effects and some of their side-effects. The inhibition of prostaglandin synthesis observed with etodolac differs from that of other NSAIDs. In an animal model at an established anti-inflammatory dose, cytoprotective PGE concentration in the gastric mucosa have been shown to be reduced to a lesser degree and for a shorter period than other NSAIDs. This finding is consistent with subsequent in-vitro studies which have found etodolac to be selective for induced cyclo-oxygenase 2 (COX-2, associated with inflammation) over COX-1 (cytoprotective).
Furthermore, studies in human cell models have confirmed that etodolac is selective for the inhibition of COX-2.
The clinical benefit of preferential COX-2 inhibition over COX-1 has yet to be proven.
Anti-inflammatory effects: Experiments have shown etodolac to have marked anti-inflammatory activity, being more potent than several clinically established NSAIDs.
Paracetamol
Analgesic – The mechanism of analgesic action has not been fully determined. Paracetamol may act predominantly by inhibiting prostaglandin synthesis in the central nervous system (CNS) and to a lesser extent, through a peripheral action by blocking pain-impulse generation. The peripheral action may also be due to inhibition of prostaglandin synthesis or to inhibition of the synthesis or actions of other substances that sensitise pain receptors to mechanical or chemical stimulation.
Antipyretic – Paracetamol probably produces anti-pyresis by acting centrally on the hypothalamic heat-regulation centre to produce peripheral vasodilation resulting in increased blood flow through the skin, sweating and heat loss. The central action probably involves inhibition of prostaglandin synthesis in the hypothalamus.
5.3 Pharmacokinetic properties
S (+) Etodolac is well absorbed and does not undergo significant first-pass metabolism following oral administration. The extent of absorption of S (+) Etodolac is not affected after a meal. Food intake, however, reduces the peak concentration reached by approximately one-half and increases the time to peak concentration.
Paracetamol is well absorbed from the gastrointestinal tract in humans. The ingestion of food along with Paracetamol does not seem to affect its absorption.
| Parameters | S (+) Etodolac | Paracetamol |
| Oral bioavailability | 80% | 63 – 89% |
| Cmax | 4.07 µg/ml | 12.3 ng/mL |
| Tmax | 3.3 hr | 1.1 hr |
| Vd | Approx. 390 mL/kg | 51 L |
| Plasma protein binding | More than 99% | 10% to 25% |
| Half-life | 6.2 hr | 2 hr |
| Metabolism | S (+) Etodolac is extensively metabolized in the liver. The metabolites include 6-, 7-, and 8-hydroxylated-etodolac and Etodolac glucuronide. | Metabolism is primarily in the liver, via conjugation with Glucuronic (60%) and Sulphuric (35%) acids, or Cysteine (3%). A small amount of drug undergoes cytochrome P-450 mediated N-hydroxylation to form N-acetylp-aminobenzoquino-neimine. |
| Excretion | Approx. 72% of the dose excreted into urine as parent drug plus metabolite. Fecal excretion accounts for 16% of the dose. | 90 - 100% of the drug is recoverable in the urine as conjugates |
6.0 Nonclinical Properties
6.1 Animal Toxicology or Pharmacology
Etodolac: Not Available
Paracetamol: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development. Conventional studies using the currently accepted standards for the evaluation of toxicity to reproduction and development are not available
7.0 Description
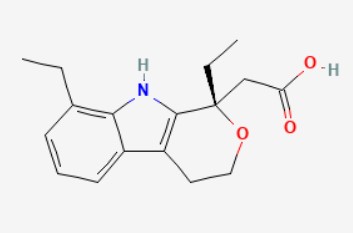
(S)-etodolac is the S-enantiomer of etodolac. It is a preferential inhibitor of cyclo-oxygenase 2 and a non-steroidal anti-inflammatory, whereas the enantiomer, (R)-etodolac, is inactive. The racemate is commonly used for the treatment of rheumatoid arthritis and osteoarthritis, and for the alleviation of postoperative pain. Chemical Name: 2-[(1S)-1,8-diethyl-4,9-dihydro-3H-pyrano[3,4-b]indol-1-yl]acetic acid Molecular Formula: C17H21NO3 Molecular Weight: 287.35 g/mol
Structure:
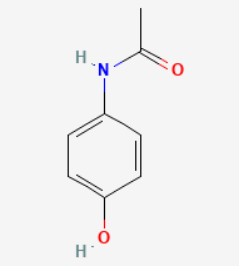
Paracetamol is a well-established analgesic.
Chemical Name: N-(4-hydroxyphenyl)acetamide
Molecular Formula: C8H9NO2
Molecular Weight: 151.2 g/mol
Structure:
8.0 Pharmaceutical particulars
8.1 Incompatibilities
None
8.2 Shelf life
Refer on the pack.
8.3 Packaging Information
PVC-Alu blister strip of 10 tablets
8.4 Storage and handling instructions
Store below 30ºC. Protect from light and moisture. Keep out of reach of children. Tablet should be swallowed whole & not to be broken, chewed or crushed.
Any unused product or waste material should be disposed of in accordance with local requirements.
9.0 Patient Counselling Information
- Take it with food to avoid getting an upset stomach.
- The drug should be taken precisely as it has been prescribed. Meant for oral consumption, Setolac®-P Tablet should be taken whole. Do not chew or crush it as it may reduce its effect on the body.
- It is best that you do not stop the medication mid-course as the pain may reoccur. Complete the entire treatment course for the best results.
- Avoid consuming alcohol when taking Setola®-P Tablet as it may cause excessive drowsiness and increase the risk of liver damage.
- Do not take it with any other medicine containing acetaminophen (drugs for pain / fever or cough-and-cold) without asking your doctor first.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
Keep this leaflet. You may need to read it again. If you have any further questions, please ask your doctor or pharmacist. This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. If you get any side effects, talk to you doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet:
1.What Setolac®-P Tablets is and what it is used for
2.What you need to know before you take Setolac®-P Tablets
3.How to take Setolac®-P Tablets
4.Possible side effects
5.How to store Setolac®-P Tablets
6.Contents of the pack and other information
1. What Setolac®-P Tablets is and what it is used for
Setolac®-P is one of a group of medicines called "non-steroidal anti-inflammatory drugs" (NSAIDs) which are usually taken to relieve the pain, stiffness, inflammation and swelling which is often associated with arthritis.
Setolac®-P Tablets is used to treat the symptoms of rheumatoid arthritis, osteoarthritis and ankylosing spondylitis by reducing inflammation, swelling, stiffness, and joint pain.
Each tablet contains 200mg of the S (+) Etodolac and 325mg of Paracetamol. S (+) Etodolac is the pharmacologically active component of racemate Etodolac. S (+) Etodolac has anti-inflammatory, analgesic, and antipyretic activities. Paracetamol works by relieving pain and reducing high temperature and fever. It is for effective relief from: mild to moderate pain including headache, migraine, sharp nerve pain (neuralgia), toothache, sore throat, period pains, aches and pain, symptomatic relief of rheumatic aches and pains, influenza symptoms, feverishness and feverish colds.
2. What you need to know before you take Setolac®-P Tablets
DO NOT take Setolac®-P Tablets if you:
- If you are allergic to etodolac or paracetamol or any of the other ingredients of this medicine (listed in section 6)
- If you have severe heart failure
- If you have a peptic ulcer (a small erosion or hole in the stomach or duodenum) or bleeding in your stomach, or have had two or more episodes of peptic ulcers, stomach bleeding or perforation.
- If you have had an allergic reaction or asthmatic type reaction (e.g., wheezing, itching or skin rash) after taking aspirin, Setolac®-P Tablets or another NSAID
- If you have hepatic failure and renal failure
- If you are pregnant, think you may be pregnant or are breast feeding
- If you have severe hepatocellular insufficiency
Warnings and Precautions
Talk to your doctor or pharmacist before taking Setolac®-P Tablets.
- If you suffer from kidney, heart or liver disease (including alcoholic liver disease), or a blood disorder, especially if you are also taking diuretics (water tablets). The dose should be as low as possible and you should have regular checks
- If you are already on long-term therapy with a medicine other than Setolac®-P Tablets, since your doctor will want to arrange regular check-ups, especially if you are elderly
- If you suffer from fluid retention (swelling of legs, ankles or feet)
- If you suffer from high blood pressure or heart failure
- If you suffer from, or have ever suffered from, asthma or breathing difficulties
- If you have heart problems, previous stroke or think that you might be at risk of these conditions (for example if you have high blood pressure, diabetes or high cholesterol or are a smoker)
- If you suffer from G-6-PD deficiency (a hereditary condition leading to low red blood cell counts).
Medicines such as Setolac®-P Tablets may be associated with a small increased risk of heart attack (myocardial infarction) or stroke. Any risk is more likely with high doses and prolonged treatment. Do not exceed the recommended dose or duration of treatment.
If you have heart problems, previous stroke or think that you might be at risk of these conditions (for example if you have high blood pressure, diabetes or high cholesterol or are a smoker) you should discuss your treatment with your doctor or pharmacist.
Serious gastrointestinal side effects such as bleeding, ulceration and perforation can occur at any time with or without warning symptoms in patients treated with NSAIDs. If any sign of gastrointestinal bleeding occurs, Setolac®-P Tablets should be stopped immediately.
Children
Setolac®-P Tablets is not recommended for use in children.
Other medicines and Setolac®-P Tablets
Tell your doctor if you are taking, have recently taken or might take any other medicines. Tell your doctor if you are taking:
- Warfarin - for blood thinning
- Cyclosporin - following transplantation
- Digoxin - for heart problems
- Lithium - for mental illness
- Methotrexate - used to treat conditions such as psoriasis or rheumatoid arthritis
- Corticosteroids (such as prednisolone)
- Quinolone antibiotics e.g. ciprofloxacin
- Aspirin
- Other NSAIDs - ibuprofen, naproxen, diclofenac
- Drugs to control high blood pressure
- Mifepristone (a drug to induce abortion) in the last 12 days
- Warfarin
- Antibiotics (chloramphenicol)
- anti-sickness treatments (metoclopramide, domperidone)
- treatments for diarrhoea caused by gall bladder disease
- lipid lowering drugs (cholestyramine).
If you have a blood or urine test, tell your doctor that you are taking Setolac®-P Tablets, as the drug may affect the results.
Pregnancy, breast-feeding and fertility
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine. You should inform your doctor if you have problems becoming pregnant. NSAIDs may make it more difficult to become pregnant.
Do not take Setolac®-P Tablets if you are in the last 3 months of pregnancy as it could harm your unborn child or cause problems at delivery. It can cause kidney and heart problems in your unborn baby. It may affect your and your baby’s tendency to bleed and cause labour to be later or longer than expected. You should not take Setolac®-P Tablets during the first 6 months of pregnancy unless absolutely necessary and advised by your doctor. If you need treatment during this period or while you are trying to get pregnant, the lowest dose for the shortest time possible should be used. If taken for more than a few days from 20 weeks of pregnancy onward, Setolac®-P Tablets can cause kidney problems in your unborn baby that may lead to low levels of amniotic fluid that surrounds the baby (oligohydramnios) or narrowing of a blood vessel (ductus arteriosus) in the heart of the baby. If you need treatment for longer than a few days, your doctor may recommend additional monitoring.
Setolac®-P Tablets should not be used if you are breast-feeding. It is not known if this medicine passes into breast milk. It is not recommended for use during breast-feeding unless considered essential by your doctor.
Driving and using machines
Setolac®-P may cause dizziness. So, you should not drive or operate heavy machinery if you feel dizzy or not fully alert.
3. How to take Setolac®-P Tablets
Always take Setolac®-P Tablets exactly as your doctor told you. Check with your doctor or pharmacist if you are not sure. Check the pharmacist's label for the dose recommended for you.
The recommended adult dose is one Setolac®-P tablet twice a day.
The tablet should be swallowed whole with a glass of water. Take with or after food. Do not chew or crush the tablet. Setolac®-P Tablets is not recommended for use in children.
If you take more Setolac®-P Tablets than you should
If you take more tablets than you should (an overdose), seek medical attention immediately. Always take the bottle (or packaging) with you, even if empty. Symptoms of overdose include headache, feeling sick, vomiting, stomach pain, passing blood in faeces or passing black tarry stools. On rare occasions diarrhoea, disorientation, excitation, coma, drowsiness, dizziness, ringing in the ears, fainting, and convulsive fitting may occur. In cases of significant overdose kidney failure and liver damage are possible. In some cases, irregular heartbeat and inflammation of the pancreas is also reported.
If you forget to take Setolac®-P Tablets
Do not take a double dose to make up for the forgotten dose. Take your tablet as soon as you remember and continue to take your medicine as usual, but do not take more than one tablet a day.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
The most serious side effects that may occur with Setolac®-P Tablets are serious allergic or hypersensitivity reactions, heart failure, stroke, kidney failure, liver failure, inflammation of the pancreas and aseptic meningitis. If you suffer from any of the symptoms described below: stop taking Setolac®-P Tablets and call a doctor straight away.
Allergic or hypersensitivity reactions may have the following symptoms: wheezing, difficulty breathing or shortness of breath, swelling of the face, lips, mouth or tongue extensive rash, peeling or blistering of the skin, continuous itching.
Heart and blood circulatory disorders symptoms: Chest pain, high blood pressure, swelling of the ankles, palpitations (throbbing of heart), several types of anaemia or other blood disorders, unexpected bruising and bleeding.
Stomach and bowel (gastrointestinal) problems: If you Pass blood in your faeces (stools/motions) Pass black tarry stools. Vomit any blood or dark particles that look like coffee grounds.
Kidney failure symptoms: Difficulty or pain when passing urine, discolouration of urine or urinating more or less often than usual.
Liver failure and inflammation of the pancreas symptoms: Jaundice (yellowing of the eyes or skin), abdominal pain, abnormal liver function test results.
Aseptic Meningitis symptoms
A serious rare condition known as aseptic meningitis may occur in patients with other auto-immune conditions such as systemic lupus erythematosus or mixed connective tissue disease. The symptoms of aseptic meningitis are: a very high temperature, being sick, a headache, a blotchy rash that does not fade when a glass is rolled over it (this may not develop), a stiff neck, a dislike of bright lights, drowsiness and fits.
Other reported side effects are:
Sensory disorders such as headache, ringing or buzzing in ears, dizziness, abnormal vision, hallucinations, tingling, pricking and burning of the skin (pins and needles) and vertigo (a sensation that objects are moving or spinning). Gastrointestinal problems such as mouth ulcers, sore mouth, nausea, vomiting, stomach upsets, diarrhoea, constipation, wind, heartburn, indigestion. Skin disorders such as swelling of tissues, itching of the skin, rash, redness. General disorders such as fever, drowsiness, tiredness, weakness, sleeplessness, shaking, nervousness, depression, confusion. Occasionally the blood does not clot well, which may result in easy bruising or bleeding. Rarely, a severe reduction in the number of white blood cells, which makes infections more likely.
Other rare side effects like shortness of breath
If any of the side effects become serious, or if you notice any side effects not listed in this leaflet, please tell your doctor or pharmacist.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician
5. How to store Setolac®-P Tablets
Keep out of reach of children. Protect from light and moisture. Setolac®-P Tablets should be kept at room temperature (below 300C).
6. Contents of the pack and other information
What Setolac®-P Tablets contains
Each uncoated tablet contains
S (+) Etodolac 200 mg
Paracetamol IP 325 mg
Excipients q.s.
What Setolac®-P Tablets looks like and contents of the pack
For More Information About This Product
Soventus®-dx Syrup
1.0 Generic Name
Chlorpheniramine Maleate & Dextromethorphan Hydrobromide syrup
2.0 Qualitative and quantitative composition
Each 5 ml Contains:
Dextromethorphan Hydrobromide IP 10 mg
Chlorpheniramine maleate IP 2 mg
Excipients q. s.
Colour: Sunset Yellow FCF
In a mentholated flavoured syrup base
3.0 Dosage form and strength
Syrup
100 ml bottle
4.0 Clinical particulars
4.1 Therapeutic Indication
For the temporary relief of cough due to throat irritation, sneezing & running nose.
4.2 Posology and method of administration
2 to 5 years of age: 2.5 ml (1/2 teaspoonful) 3 to 4 times a day.
6 to 12 years of age: 5 ml (1 teaspoonful) 3 to 4 times a day.
Adult (> 12 years of age): 10 ml (2 teaspoonful) 3 to 4 times a day.
4.3 Contraindications
- Hypersensitivity to Chlorpheniramine, Dextromethorphan or to any component of the formulation.
- Receiving monoamine oxidase inhibitors [MAOIs] (drugs for depression, psychiatric or emotional conditions, or Parkinson's disease) with or within 2 weeks.
- Patients with risk of developing respiratory failure
- Acute Asthma
- Patient with liver disease
- Persistent or chronic productive cough
4.4 Special warnings and precautions for use
Chlorpheniramine maleate
This medicine should be given with caution to patients with epilepsy, severe cardiovascular disorders, liver disorders, renal impairment, glaucoma, urinary retention, prostatic enlargement, pyloroduodenal obstruction, asthma, bronchitis, bronchiectasis, thyrotoxicosis and severe hypertension.
Special care should be taken when using chlorpheniramine maleate in children and the elderly as they are more likely to experience the neurological anticholinergic effects and paradoxical excitation (e.g. Increased energy, restlessness, nervousness). Avoid use in elderly patients with confusion.
The anticholinergic properties of chlorphenamine may cause drowsiness, dizziness, blurred vision and psychomotor impairment in some patients which may seriously affect ability to drive and use machinery. The effects of alcohol may be increased and therefore concurrent use should be avoided.
Should not be used with other antihistamine containing products, including antihistamine containing cough and cold medicines.
Concurrent use with drugs which cause sedation such as anxiolytics and hypnotics may cause an increase in sedative effects, therefore medical advice should be sought before taking chlorphenamine concurrently with these medicines.
Dextromethorphan Hydrobromide
Dextromethorphan should not be given to patients at risk of developing respiratory failure. Caution is needed in patients with a history of asthma and it should not be given during an acute attack. Care is also advisable in patients with bronchitis, emphysema, or in other conditions where chronic or persistent cough occurs.
4.5 Drugs interactions
Chlorpheniramine maleate
This medicine may enhance the sedative effects of alcohol, hypnotics, anxiolytics, sedatives, opioid analgesics and neuroleptics.
The anti-muscarinic effects of chlorpheniramine are enhanced by other anti-muscarinic drugs and both anti-muscarinic and sedative effects are enhanced by monoamine oxidase inhibitors and tricyclic antidepressants.
Metabolism of phenytoin may be inhibited by chlorpheniramine with the possible development of phenytoin toxicity.
Dextromethorphan Hydrobromide
Monoamine Oxidase Inhibitors (MAOIs): Dextromethorphan should not be used concurrently in patients taking monoamine oxidase inhibitors (MAOIs) or within 14 days of stopping treatment with MAOIs as there is a risk of serotonin syndrome (pyrexia, hallucinations, gross excitation or coma, hypertension, arrhythmias).
CYP2D6 inhibitors: Dextromethorphan is metabolized by CYP2D6 and has an extensive first-pass metabolism. Concomitant use of potent CYP2D6 enzyme inhibitors can increase the dextromethorphan concentrations in the body to levels multi-fold higher than normal. This increases the patient's risk for toxic effects of dextromethorphan (agitation, confusion, tremor, insomnia, diarrhoea and respiratory depression) and development of serotonin syndrome. Potent CYP2D6 enzyme inhibitors include SSRIs such as fluoxetine and paroxetine, quinidine and terbinafine. In concomitant use with quinidine, plasma concentrations of dextromethorphan have increased up to 20-fold, which has increased the CNS adverse effects of the agent. Amiodarone, flecainide and propafenone, sertraline, bupropion, methadone, cinacalcet, haloperidol, perphenazine and thioridazine also have similar effects on the metabolism of dextromethorphan. If concomitant use of CYP2D6 inhibitors and dextromethorphan is necessary, the patient should be monitored and the dextromethorphan dose may need to be reduced.
CNS depressants: Dextromethorphan might exhibit additive CNS depressant effects when co-administered with alcohol, antihistamines, psychotropics, and other CNS depressant drugs.
4.6 Use in special populations
Pregnant Women
There are no adequate and well-controlled studies of this combination in pregnant women. There are no adequate controlled studies of chlorpheniramine in pregnant women and therefore should not be used during pregnancy. There are no adequate and well-controlled studies of Dextromethorphan in pregnant women. Dextromethorphan should not be used during pregnancy or lactation unless the potential benefit of treatment to the mother outweighs the possible risk to the developing foetus or nursing infant.
Caution is advised when Soventus®-DX Syrup is used during pregnancy.
Lactating Women
Chlorpheniramine may be secreted in breast milk. Hence, it is not recommended in nursing mothers because of the risk of adverse effects, such as unusual excitement or irritability in infants. Chlorpheniramine may also inhibit lactation. It is not known whether dextromethorphan or its metabolites are excreted in breast milk. A decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Paediatric Patients
Safety and efficacy of this formulation in neonates and children below 2 years of age has not been established. Thus, Soventus®-DX Syrup is not recommended for use in paediatric patients below 2 years of age.
Geriatric Patients
The elderly patients taking Chlorpheniramine are more likely to experience neurological anticholinergic effects. Elderly patients with normal renal and hepatic function may be given the same dose as recommended for adults. Elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Renal Impairment Patients
Caution should be exercised while using Soventus®-DX syrup in patients with significant renal dysfunction. Dose should be reduced or the dosing interval must be extended in patients with severe renal impairment.
Hepatic Impairment Patients
In hepatic impairment, Soventus®-DX syrup should be used with caution.
4.7 Effects on ability to drive and use machines
Soventus®-DX Syrup may cause side effects which could affect your ability to drive. Chlorpheniramine may cause blurred vision, dizziness, drowsiness and interfere with human performance and therefore may seriously influence the ability to drive and operate machinery. Dextromethorphan can impair cognitive function and can affect a patient's ability to drive safely.
4.8 Undesirable Effects
Chlorpheniramine maleate
Adverse reactions identified during post-marketing use with chlorphenamine are listed below. As these reactions are reported voluntarily from a population of uncertain size, the frequency of some reactions is unknown but likely to be rare or very rare:
Children and the elderly are more susceptible to neurological anticholinergic effects and paradoxical excitation (e.g. increased energy, restlessness, nervousness)
Dextromethorphan Hydrobromide
ADRs are presented by frequency category based on 1) incidence in adequately designed clinical trials or epidemiology studies, if available, or 2) when incidence cannot be estimated, frequency category is listed as 'Not known’.



frequency category is listed as 'Not known’.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit / risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website : http://www.zuventus.co.in/safety.aspx
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
The estimated lethal dose of chlorphenamine is 25 to 50mg/kg body weight. Symptoms and signs include sedation, paradoxical excitation of the CNS, toxic psychosis, convulsions, apnoea, anticholinergic effects, dystonic reactions and cardiovascular collapse including arrhythmias. Symptomatic and supportive measures should be provided with special attention to cardiac, respiratory, renal and hepatic functions and fluid and electrolyte balance. If overdosage is by the oral route, treatment with activated charcoal should be considered provided there are no contraindications for use and the overdose has been taken recently (treatment is most effective if given within an hour of ingestion.) Treat hypotension and arrhythmias vigorously. CNS convulsions may be treated with i.v. diazepam. Haemoperfusion may be used in severe cases.
Dextromethorphan is thought to be of low toxicity, but the effects in overdosage will be potentiated by simultaneous ingestion of alcohol and psychotropic drugs. Symptoms of overdose may include mydriasis, nausea and vomiting, CNS depression, excitation, lethargy, nystagmus, psychomotor hyperactivity, serotonin syndrome, somnolence (drowsiness), dizziness, dysarthria (slurred speech), mental confusion, psychotic disorder (psychosis), and respiratory depression. Treatment should be symptomatic and supportive. Gastric lavage may be of use. Naloxone has Patients should be kept under observation and symptomatic and supportive treatment is advised. been used successfully to reverse central or peripheral opioid effects of dextromethorphan in children (0.01mg/kg body weight).
5.0 Pharmacological properties
5.1 Mechanism of Action
Chlorpheniramine maleate
Chlorpheniramine is a first-generation alkylamine antihistamine. It is a less sedating H1 blocker with autonomic effects (anti-cholinergic action), used in the prevention of the symptoms of allergic conditions such as rhinitis.
Dextromethorphan Hydrobromide
Dextromethorphan (D-3-methoxy-N-methylmorphinan) is the D-isomer of the codeine analog methorphan. It is an antitussive drug and one of the active ingredients used to prevent coughs.
5.2 Pharmacodynamic properties
Chlorpheniramine maleate
Chlorphenamine is a potent antihistamine (H1-antagonist). Antihistamines diminish or abolish the actions of histamine in the body by competitive reversible blockade of histamine H1-receptor sites on tissues. Chlorphenamine also has anticholinergic activity. Antihistamines act to prevent the release of histamine, prostaglandins and leukotrienes and have been shown to prevent the migration of inflammatory mediators. The actions of chlorphenamine include inhibition of histamine on smooth muscle, capillary permeability and hence reduction of oedema and wheal in hypersensitivity reactions such as allergy and anaphylaxis.
Dextromethorphan Hydrobromide
Dextromethorphan is the dextrorotatory isomer of 3-methoxy-N-methyl-morphinan. It is a synthetic morphine derivative that, in contrast to its levo-rotatory isomer, has no significant analgesic, respiratory depressant or physical dependency properties at recommended doses.
Dextromethorphan is a non-opioid antitussive drug. It exerts its antitussive activity by acting on the cough centre in the medulla oblongata, raising the threshold for the cough reflex. The onset of antitussive effects are realised within 15 to 30 minutes of oral administration, lasting for approximately 3 to 6 hours.
The major metabolite of dextromethorphan, dextrorphan, binds with high affinity to σ-receptors to produce its antitussive activity without exhibiting the classic opiate effects that occur from binding into μ- and δ-receptors. Dextrorphan also exhibits binding activity at serotonergic receptors and was shown to enhance serotonin activity by inhibiting the reuptake of serotonin. In larger than therapeutic doses, dextrorphan is also an antagonist of N-methyl-D-aspartate (NMDA) receptors.
5.3 Pharmacokinetic properties
Chlorpheniramine maleate
Chlorpheniramine maleate is almost completely absorbed after administration by mouth, peak plasma concentrations occurring at about 2.5 to 6 hours. The drug is widely distributed including passage into the CNS, with a volume of distribution of between 1 and 10L/KG. About 70% of chlorpheniramine in the circulation is protein-bound. Chlorpheniramine undergoes some first pass metabolism and enterohepatic recycling. Chlorpheniramine is extensively metabolised, principally to inactive desmethylated metabolites which are excreted primarily in the urine, together with about 35% unchanged drug. Only trace amounts are excreted in the faeces. The mean elimination half-life has been reported to be about 30 hours, with mean values ranging from 2 to 43 hours.
Dextromethorphan Hydrobromide
Absorption: Dextromethorphan is rapidly absorbed from the gastrointestinal tract with peak plasma concentrations reached in approximately 2 to 2.5 hours. The low plasma levels of dextromethorphan suggest low oral bioavailability secondary to extensive first-pass (pre-systemic metabolism) in the liver. The maximum clinical effects occur 5 to 6 hours after ingestion of dextromethorphan.
Distribution: Dextromethorphan is widely distributed in the human body. Dextromethorphan and its active metabolite, dextrorphan, are actively taken up and concentrated in brain tissue. It is not known if dextromethorphan or dextrorphan are excreted in breast milk or cross the placenta.
Metabolism: Dextromethorphan undergoes rapid and extensive first-pass metabolism in the liver after oral administration. Genetically controlled O-demethylation (CYD2D6) is the main determinant of dextromethorphan pharmacokinetics in human volunteers. It appears that there are distinct phenotypes for this oxidation process resulting in highly variable pharmacokinetics between subjects. Unmetabolised dextromethorphan, together with the three demethylated morphinan metabolites dextrorphan (also known as 3-hydroxy-N-methylmorphinan), 3- hydroxymorphinan and 3-methoxymorphinan have been identified as conjugated products in the urine. Dextrorphan, which also has antitussive action, is the main metabolite. In some individuals metabolism proceeds more slowly and unchanged dextromethorphan predominates in the blood and urine.
Excretion: Dextromethorphan is primarily excreted via the kidney as unchanged parent drug and its active metabolite, dextrorphan. Dextrorphan and 3-hydroxy-morphinan are further metabolised by glucuronidation and are eliminated via the kidneys. The elimination half-life of the parent compound is between 1.4 to 3.9 hours; dextrorphan is between 3.4 to 5.6 hours. The half-life of dextromethorphan in poor metabolisers is extremely prolonged, in the range of 45 hours.
6.0 Nonclinical Properties
6.1 Animal Toxicology or Pharmacology
Chlorpheniramine maleate
The antihistaminic potency of chlorpheniramine is confined mainly to its (+)-isomer. The racemate is similarly or slightly more toxic because of the contribution of (-)-isomer. The toxicity may therefore be non-specific, perhaps attributable to local anaesthetic action and the toxic effects (excitation/sedation, coma, convulsions and death) resemble those of other classic H1antihistamines. Toxic doses may cause hypotension attributable to myocardial depression, an effect which is clearer with the (-)-isomer. The experimental data on the carcinogenicity and mutagenicity of chlorpheniramine indicate lack of these adverse effects, but the racemate and the (+)-isomer have shown some embryotoxicity in fertility tests. Effective antihistaminic concentrations of chlorpheniramine in vitro are about 1-10µg/L and oral doses of 0.2-1 mg/kg antagonise histamine-induced bronchospasm in guinea pigs.
Dextromethorphan Hydrobromide
General toxicology: Acute oral toxicity studies conducted with Dextromethorphan report the following LD50 values (mg/kg): mouse, 210 and rat, 116. Acute subcutaneous toxicity with Dextromethorphan reports the LD50 value (mg/kg): mouse, 112. Acute intravenous toxicity with Dextromethorphan reports the LD50 value (mg/kg): rat, 16.3.
Repeat dose toxicity studies conducted in rats for 13-week duration at doses up to 100 mg/kg and 27 weeks at 10 mg/kg, and of 14 weeks in dogs by oral gavage at doses up to 4 mg/kg on five days per week. The only effect recorded was of reduced body weight gain in the rat 13-week study at the highest dose.
Genetic Toxicology: Dextromethorphan hydrobromide was negative in the bacterial reverse mutation assay (Ames test). Dextromethorphan 39 mg/kg is reported to be negative in in-vivo mouse micronucleus test and comet assay. Dextromethorphan was reported to be negative in in vitro chromosome aberration assay tested up to 200 μg/ml.
Carcinogenicity: There are no known reports of animal carcinogenicity studies for Dextromethorphan. The overall weight of evidence for Dextromethorphan and its structural analogues, support the conclusion that this class of phenanthrene-based chemicals, and Dextromethorphan, in particular, are not genotoxic in vitro or in vivo
Teratogenicity: There was no association between dextromethorphan and malformations.
Fertility: Mating, gestation, fertility, littering and lactation were studied in rats at doses up to 50 mg/kg and no adverse effects were found.
7.0 Description
Each 5 ml of Soventus®-DX syrup contains 10 mg of Dextromethorphan hydrobromide and 2 mg of chlorpheniramine maleate for oral administration.
Chlorpheniramine maleate
Chlorphenamine is a potent antihistamine (H1-antagonist). Antihistamines diminish or abolish the actions of histamine in the body by competative reversible blockade of histamine H1-receptor sites on tissues. Chlorphenamine also has anticholinergic activity. Molecular Weight: 390.9 g/mol Molecular Formula: C16H19ClN2.C4H4O4 Chemical Name: (Z)-but-2-enedioic acid;3-(4-chlorophenyl)-N,N-dimethyl-3-pyridin-2-ylpropan-1-amine
Dextromethorphan Hydrobromide
Dextromethorphan is the dextrorotatory isomer of 3-methoxy-N-methyl-morphinan. It is a synthetic morphine derivative. Dextromethorphan is a non-opioid antitussive drug. It exerts its antitussive activity by acting on the cough centre in the medulla oblongata, raising the threshold for the cough reflex.
Molecular Weight: 352.3 g/mol
Molecular Formula: C18H26BrNO
Chemical Name:
(1S,9S,10S)-4-methoxy-17-methyl-17-azatetracyclo[7.5.3.01,10.02,7]heptadeca-2(7),3,5-triene;hydrobromide
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf life
Refer on Pack
8.3 Packaging information
100 ml bottle
8.4 Storage and handling information
Store in a cool & dark place. Keep out of reach of children
9.0 Patient Counselling Information
- Take exactly as directed by your doctor or on the label. Do not increase the dosage or take for longer than is recommended. Use a proper measuring cup or spoon to measure the dosage.
- If you are drowsy after taking Soventus®-DX Syrup, you should not drive or operate any machinery.
- Inform your doctor if you have a history of stomach ulcers or asthma.
- Inform your doctor if you are taking any other medications, e.g., anti-depressants.
- Consult your doctor if you do not see any improvement and have a cough for > 7 days.
- Avoid consuming alcohol while taking Soventus DX Syrup as it can cause excessive drowsiness.
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet.
What is in this leaflet
1.What Soventus®-DX Syrup is and what it is used for
2.What you need to know before you take Soventus®-DX Syrup
3.How to take Soventus®-DX Syrup
4.Possible side effects
5.How to store Soventus®-DX Syrup
6.Contents of the pack and other information
1. What Soventus®-DX Syrup is and what it is used for
Soventus®-DX Syrup is a combination of Chlorpheniramine maleate & Dextromethorphan Hydrobromide. This medicine is used to help relieve dry, irritating coughs.
Dextromethorphan hydrobromide which is an antitussive to help stop persistent dry coughing. It works by reducing the activity of cough center in the brain. The other active ingredient is chlorphenamine maleate, an antihistamine which can help to relieve the symptoms of allergic reactions. It relieves allergic symptoms like runny nose, watery eyes, sneezing, throat irritation.
2. What you need to know before you take Soventus®-DX Syrup Do not take Soventus®-DX Syrup:
- If you are allergic to any of the ingredients of this medicine.
- If you are taking, or have taken in the last two weeks, drugs for depression known as Monoamine Oxidase Inhibitors (MAOIs).
- if you are taking selective serotonin re-uptake inhibitors (used to treat depression and anxiety such as fluoxetine, paroxetine and sertraline).
- If you have an acute asthma
- If you are taking other medicines containing antihistamines, including products for the relief of colds and coughs
- If you suffer from lung disease.
Warnings and precautions
Talk to your doctor, pharmacist or nurse before taking Soventus®-DX Syrup.
- If you suffer from epilepsy, heart or circulatory disease, liver problems, kidney disease.
- If you high blood pressure or glaucoma.
- If you have chronic bronchitis, emphysema or asthma or have had a cough for a few weeks or a cough with a lot of mucus (phlegm).
- If you are taking any other cough and cold medicines.
- If your child is prone to developing certain allergic reactions (e.g. atopic reactions).
- If you are or have ever been addicted to opioids, alcohol, prescription medicines, or illegal drugs.
- If you have previously suffered from withdrawal symptoms such as agitation, anxiety, shaking or sweating, when you have stopped taking alcohol or drugs.
- If you have been told by your doctor that you are a slow metabolizer of CYP2D6.
- If you are taking medicines such as certain antidepressants or antipsychotics this medicine may interact with these medicines and you may experience mental status changes (e.g. agitation, hallucinations, coma), and other effects such as body temperature above 38°C, increase in heart rate, unstable blood pressure, and exaggeration of reflexes, muscular rigidity, lack of coordination and/or gastrointestinal symptoms (e.g. nausea, vomiting, diarrhoea).
- If you are taking any other medicines, including: Certain drugs for depression such as norepinephrine-dopamine reuptake inhibitors (NDRI), which include bupropion.
- If you are taking Antipsychotics (drugs used to treat mood disorders such as haloperidol, thioridazine, perphenazine).
- If you are taking anti-arrhythmic agents (drugs used to treat an irregular heartbeat such as amiodarone, propafenone, quinidine and flecainide).
- If you are taking calcimimetic agents (drugs used to treat secondary hyperparathyroidism, elevated parathyroid hormone levels such as cinacalcet).
- Antifungals (terbinafine).
- If you are taking opioid analgesics (drugs used to relieve pain e.g. codeine, tramadol, morphine, methadone). If using antihistamines (drugs used to treat the symptoms of allergic reactions).
- If you have an overactive thyroid.
- If you have difficulty passing urine.
- If you have an obstruction in their intestine.
- If you have a rare blood disease called porphyria.
If any of these points apply to you now or in the past, talk to a doctor or pharmacist.
Tell your doctor immediately if during treatment you suffer from
hypersensitivity reactions (itchiness, redness of the skin, hives, swelling of the face, lips, mouth, tongue or throat)
Other medicines and Soventus®-DX Syrup
Tell your doctor if you are taking or have taken the following medications.
Chlorpheniramine may enhance the sedative effects of alcohol, hypnotics, anti- anxiety, sedatives, opioid analgesics and neuroleptics.
Dextromethorphan should not be used concurrently in patients taking monoamine oxidase inhibitors (MAOIs) or within 14 days of stopping treatment with MAOIs as there is a risk of serotonin syndrome (pyrexia, hallucinations, gross excitation or coma, hypertension, arrhythmias).
Dextromethorphan might exhibit additive CNS depressant effects when co-administered with alcohol, antihistamines, psychotropics, and other CNS depressant drugs.
Pregnancy, breast-feeding
Do not take this medicine, if you are pregnant or breast-feeding, think you may be pregnant or planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
Geriatric Population
Talk to your doctor or pharmacist before you take this syrup as you may be more likely to get side effects including confusion and you may need to take a lower daily dose.
Pediatric population
Not recommended for children below 2 years of age.
Renal Insufficiency
Caution should be exercised while using Soventus®-DX syrup in patients with significant renal dysfunction. Dose should be reduced or the dosing interval must be extended in patients with severe renal impairment.
Hepatic Insufficiency
Caution should be exercised in patients with moderate to severe and liver disease.
Driving and using machines
This medicine can affect your ability to drive. Do not drive whilst taking this medicine until you know how this medicine affects you. It may be an offence to drive when taking this medicine if your ability to drive safely is affected.
3. How to take Soventus®-DX Syrup
Check the seal is not broken before first use. If it is, do not give the medicine. Always use the measuring cup supplied with the pack.
2 to 5 years of age: 2.5 ml (1/2 teaspoonful) 3 to 4 times a day. 6 to 12 years of age: 5 ml (1 teaspoonful) 3 to 4 times a day. Adult (> 12 years of age): 10 ml (2 teaspoonful) 3 to 4 times a day. Do not take more than 4 doses in 24 hours. If symptoms persist or worsen, talk to your doctor or pharmacist.
If you take more Soventus®-DX Syrup than you should
If anyone has too much intake, contact a doctor or your nearest accident and emergency department (casualty) taking this leaflet and pack with you. If you take more of this medicine than you should, you may experience the following symptoms: nausea and vomiting, involuntary muscle contractions, agitation, confusion, somnolence, disturbances in consciousness, involuntary and rapid eye movements, cardiac disorders (rapid heart beating), coordination disorders, psychosis with visual hallucinations, and hyperexcitability. Also, other types of hallucinations, psychotic disorders, seizures, clumsiness, dizziness, speech problems, lack of energy, high blood pressure, tremor, or constricted or dilated pupils. Tell your doctor or emergency department in hospital immediately.
If you forget to take Soventus®-DX Syrup
If you forget a dose, do not worry. Just wait until the next dose is due. Do not take a double dose to make up for a forgotten dose.
If you stop taking Soventus®-DX Syrup
Unless your doctor instructs you to stop treatment, it is important to continue taking Soventus®-DX Syrup. If you stop and your original symptoms come back tell your doctor or pharmacist immediately.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them Tell your doctor, nurse or pharmacist immediately if you notice any of these side effects during your treatment with Soventus®-DX Syrup:
- Drowsiness and sedation are very common side effects with chlorpheniramine. Other common side effects include disturbance in concentrating, un-coordination, dizziness, headaches, blurred vision, feeling or being sick, dry mouth, fatigue.
- Dextromethorphan may cause side effect such as swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing, fits.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top end of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store Soventus®-DX Syrup
Store in a cool & dark place. Keep out of reach of children. Do not freeze.
6. Contents of the pack and other information
What Soventus®-DX Syrup contains:
Each 5 ml Contains: Dextromethorphan Hydrobromide IP 10 mg
Chlorpheniramine maleate IP 2 mg
Excipients q. s.
Colour: Sunset Yellow FCF In a mentholated flavoured syrup base
Pack size: 100 ml Bottle
For More Information About This Product
Vitanova-D3 Sachet
1. Generic Name
Cholecalciferol Granules
2. Qualitative and quantitative composition
Each sachet of 1gm contains:
Cholecalciferol IP 60000 IU.
Excipients q.s.
3. Dosage form and strength
Granules, 60000 IU per sachet
4. Clinical particulars
4.1 Therapeutic indication
For the treatment of Vitamin-D3 deficiency.
4.2 Posology and method of administration
1/4 to 1 sachet with milk or as directed by the physician
4.3 Contraindications
- Hypersensitivity to vitamin D or any of the excipients in the product
- Hypervitaminosis D
- Nephrolithiasis
- Diseases or conditions resulting in hypercalcaemia and/or hypercalciuria
- Severe renal impairment
4.4 Special warnings and precautions for use
- Patients with gastrointestinal malabsorption syndromes may require higher doses of vitamin D3 supplementation and measurement of 25-hydroxyvitamin D should be considered.
- Vitamin D3 supplementation may worsen hypercalcemia and/or hypercalciuria when administered to patients with diseases associated with unregulated overproduction of 1,25 dihydroxyvitamin D (e.g., leukaemia, lymphoma, sarcoidosis). Urine and serum calcium should be monitored in these patients.
- It should be used with caution in patients with renal impairment or calculi, or heart disease, who might be at increased risk of organ damage if hypercalcaemia occurred.
- Plasma phosphate concentrations should be controlled during vitamin D3 therapy to reduce the risk of ectopic calcification.
- It is advised that patients receiving pharmacological doses of vitamin D3 should have their plasma-calcium concentration monitored at regular intervals, especially initially or if symptoms suggest toxicity.
4.5 Drugs interactions
- Olestra, mineral oils, orlistat, and bile acid sequestrants (e.g., cholestyramine, colestipol) may impair the absorption of vitamin D3.
- Anticonvulsants, cimetidine, and thiazides may increase the catabolism of vitamin D.
- There is an increased risk of hypercalcaemia if vitamin D3 is given with thiazide diuretics, calcium, or phosphate. Plasma-calcium concentrations should be monitored in such situations.
- Some antiepileptics may increase vitamin D3 requirements (e.g. carbamazepine, phenobarbital, phenytoin, and primidone).
- Rifampicin and isoniazid may reduce the effectiveness of vitamin D3.
- Corticosteroids may counteract the effect of vitamin D3.
4.6 Use in special populations
Pregnancy
No data are available for cholecalciferol (vitamin D3). There are no studies in pregnant women. It should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and fetus.
Nursing Mothers
Cholecalciferol and some of its active metabolites pass into breast milk. Infants should be closely monitored for hypercalcemia or clinical manifestations of vitamin D toxicity if the mother is taking pharmacological doses of vitamin D3.
Infants
Vitamin D3 should be used with caution in infants, who may have increased sensitivity to its effects.
4.7 Effects on ability to drive and use machines
It is not expected that Vitanova sachet would affect your ability to drive or to operate machinery.
4.8 Undesirable effects
Major & minor side effects for Vitanova D3 Sachet
- Increased blood calcium levels
- Increased calcium levels in urine
- Constipation
- Skin rash, hives, or itching
- Chest pain
- Shortness of breath
Reporting of suspected adverse reactions
- Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
- Website: http://www.zuventus.co.in/safety.aspx
- By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
There is limited information regarding doses of cholecalciferol associated with acute toxicity, although intermittent (yearly or twice yearly) single doses of ergocalciferol (vitamin D2) as high as 600,000 IU have been given without reports of toxicity. Signs and symptoms of vitamin D toxicity include hypercalcemia, hypercalciuria, anorexia, nausea, vomiting, polyuria, polydipsia, weakness, and lethargy. Serum and urine calcium levels should be monitored in patients with suspected vitamin D toxicity.
Treatment
Standard therapy includes restriction of dietary calcium, hydration, and systemic glucocorticoids in patients with severe hypercalcemia. Dialysis to remove vitamin D would not be beneficial.
5. Pharmacological properties
5.1 Mechanism of Action
The mechanism of action of 1,25(OH)2D(calcitriol) is mediated by the interaction of calcitriol with the vitamin D receptor (VDR). Calcitriol binds to cytosolic VDRs within target cells, and the receptor-hormone complex translocates to the nucleus and interacts with DNA to modify gene transcription. The VDR belongs to the steroid and thyroid hormone receptor supergene family. Calcitriol also exerts nongenomic effects that may require the presence of a functional VDR.
5.2 Pharmacodynamic properties
Vitamin D3 is converted to 25-hydroxyvitamin D3 in the liver. Conversion to the active calcium-mobilizing hormone 1, 25-dihydroxyvitamin D3 (calcitriol) in the kidney is stimulated by both parathyroid hormone and hypophosphatemia. The principal action of 1,25-dihydroxyvitamin D3 is to increase intestinal absorption of both calcium and phosphate as well as regulate serum calcium, renal calcium and phosphate excretion, bone formation and bone resorption.
Vitamin D is required for normal bone formation. Vitamin D insufficiency develops when both sunlight exposure and dietary intake are inadequate. Insufficiency is associated with negative calcium balance, increased parathyroid hormone levels, bone loss, and increased risk of skeletal fracture. In severe cases, deficiency results in more severe hyperparathyroidism, hypophosphatemia, proximal muscle weakness, bone pain and osteomalacia.
5.3 Pharmacokinetic properties
Absorption
Vitamin D substances are well absorbed from the gastrointestinal tract. The presence of bile is essential for adequate intestinal absorption; absorption may be decreased in patients with decreased fat absorption.
Distribution
Following absorption, vitamin D3 enters the blood as part of chylomicrons. Vitamin D3 is rapidly distributed mostly to the liver where it undergoes metabolism to 25-hydroxyvitamin D3, the major storage form. Lesser amounts are distributed to adipose tissue and stored as vitamin D3 at these sites for later release into the circulation. Circulating vitamin D3 is bound to vitamin D-binding protein.
Metabolism
Vitamin D3 is rapidly metabolized by hydroxylation in the liver to 25-hydroxyvitamin D3, and subsequently metabolized in the kidney to 1,25-dihydroxyvitamin D3, which represents the biologically active form. Further hydroxylation occurs prior to elimination. A small percentage of vitamin D3 undergoes glucuronidation prior to elimination.
Excretion
When radioactive vitamin D3 was intravenously administered to healthy subjects, the mean urinary excretion of radioactivity after 48 hours was 2.4% of the administered dose, and the mean fecal excretion of radioactivity after 48 hours was 4.9% of the administered dose. In both cases, the excreted radioactivity was almost exclusively as metabolites of the parent. The mean half-life of vitamin D3 in the serum following an oral dose is approximately 14 hours.
6. Nonclinical properties
6.1 Animal Toxicology or Pharmacology
No known animal toxicology data
7. Description
Vitanova D3 contains cholecalciferol (Vitamin D3). Vitamin D3 is essential for the proper growth and development of the body. It is synthesized within the body after exposure to sunlight and is essential for many important functions of the human body. Vitamin D3 in Vitanova D3 also increases the Calcium absorption from the intestines.
8. Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on pack
8.3 Packaging information
4 sachets of 1 gram each
8.4 Storage and handing instructions
Store protected from light & moisture at a temperature not exceeding 25°C. Keep out of reach of children.
9. Patient Counselling Information
- Take exactly as directed by your doctor or on the label. Do not increase the dosage or take for longer than is recommended.
- Vitanova-D3 Sachets 1 gm may interfere with cholesterol tests, hence please inform your physician and laboratory staff that you are taking Vitanova-D3 Sachets 1 gm before undergoing blood tests.
- Clinical monitoring of serum electrolyte concentrations and cardiac function is recommended.
About leaflet
Read all of this leaflet carefully before you start using this medicine, because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist, or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, or pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What VITANOVA-D3 is and what it is used for
- What you need to know before you use VITANOVA-D3
- How to use VITANOVA- D3
- Possible side effects
- How to store VITANOVA- D3
- Contents of the pack and other information
1. What VITANOVA- D3 is and what it is used for
VITANOVA- D3 sachet contains the active ingredient cholecalciferol granules (vitamin D3). Vitamin-D3, can be found in some foods and is also produced by the body when skin is exposed to sunlight. Vitamin D3 helps the kidneys and intestine absorb calcium and it helps build bones. Vitamin D3 deficiency is the predominant cause of rickets (defective mineralization of bones in children) and osteomalacia (inadequate mineralization of bones in adults). Vitamin D3 is essential for the growth, formation, and maintenance of healthy bones. Also Vitamin D also regulates many other cellular functions in your body. Its anti-inflammatory, antioxidant and neuroprotective properties support immune health, muscle function and brain cell activity.
Vitanova -D3 Sachet is a vitamin D3 supplement which is primarily used to manage and prevent vitamin D3 deficiency states in adults and children.
2. What you need to know before you use VITANOVA- D3
Do not use VITANOVA- D3:
- if you are allergic to vitamin D3 or any of the other ingredients of this medicine;
- if you have high levels of calcium in your blood (hypercalcaemia) or urine (hypercalciuria);
- if you have kidney stones (renal calculi) or severe renal impairment;
- if you have high levels of vitamin D3 in your blood (hypervitaminosis D)
Warnings and precautions
Talk to your doctor, pharmacist or nurse before using VITANOVA- D3 if you:
- are undergoing treatment with certain medicines used to treat heart disorders (e.g., cardiac glycosides, such as digoxin);
- have sarcoidosis (an immune system disorder which may cause increased levels of vitamin D3 in the body);
- are treated with diuretics (e.g. benzothiadiazine)
- are immobilized
- suffer from pseudo hypoparathyroidism
- are taking medicines containing vitamin D3, or eating foods or milk enriched with vitamin D3;
- are likely to be exposed to a lot of sunshine whilst using VITANOVA- D3;
- take additional supplements containing calcium. Your doctor will monitor your blood levels of calcium to make sure they are not too high whilst you are using VITANOVA- D3;
- have kidney damage or disease and if you have a tendency for the formation of renal stones. Your doctor may want to measure the levels of calcium in your blood or urine.
- take a daily dose of vitamin D3 exceeding 1,000 I.U. over a long period of time, your doctor should monitor the level of calcium in your blood by lab test.
Other medicines and VITANOVA- D3
Tell your doctor or pharmacist if you are using, have recently used or might use any other medicines. This is especially important if you are taking:
- medicines that act on the heart or kidneys, such as cardiac glycosides (eg, digoxin) or diuretics (eg, bendroflumethiazide). When used at the same time as vitamin D3 these medicines may cause a large increase in the level of calcium in the blood and urine;
- medicines containing vitamin D3 or eating food rich in vitamin D3, such as, some types of vitamin D3-enriched milk;
- actinomycin (a medicine used to treat some forms of cancer) and imidazole antifungals (e.g., clotrimazole and ketoconazole, medicines used to treat fungal disease). These medicines may interfere with the way your body process vitamin D3;
- medicines to treat tuberculosis e.g. rifampicin, isoniazid;
- the following medicines because they can interfere with the effect or the absorption of vitamin D3:
- antiepileptic medicines (anticonvulsants), barbiturates;
- glucocorticoids (steroid hormones such as hydrocortisone or prednisolone). These can decrease the effect of vitamin D3;
- medicines that lower the level of cholesterol in the blood (such as cholestyramine, or colestipol);
- certain medicines for weight loss that reduce the amount of fat your body absorbs (e.g., orlistat);
- certain laxatives (such as liquid paraffin).
VITANOVA- D3 with food, drink and alcohol
You should take this medicine preferably together with a meal to help your body absorb the vitamin D3.
Pregnancy, breast-feeding and fertility
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine. This high strength formulation is not recommended for use in pregnant and breastfeeding women.
Driving and using machines
It is not expected that Vitanova-D3 sachet would affect your ability to drive or to operate machinery.
3. How to use VITANOVA- D3
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure. You should take VITANOVA- D3 sachet preferably together with a meal.
Use in adults and Children
Pour the contents of the sachet into a cup of milk, stir, then drink immediately. The recommended dosage is one fourth to one sachet per day. Your doctor or pharmacist will tell you how many sachets you need to take a day. This initial treatment may be followed by maintenance therapy, as directed by your doctor.
Your doctor will adjust the dose for you.
If you take more VITANOVA- D3 than you should
If you take more medicine than prescribed, stop using this medicine and contact your doctor. If it is not possible to talk to a doctor go to the nearest hospital emergency department and take the medicine package with you. The most common symptoms of overdose are: nausea, vomiting, excessive thirst, the production of large amounts of urine over 24 hours, constipation and dehydration, high levels of calcium in the blood and in urine (hypercalcaemia and hypercalciuria) shown by lab test.
If you forget to take VITANOVA- D3
If you forget to take a dose of VITANOVA- D3, take the forgotten dose as soon as possible. Then take the next dose at the correct time. However, if it is almost time to take the next dose, do not take the dose you have missed; just take the next dose as normal. Do not take a double dose to make up for a forgotten dose. If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Possible side effects may include:
Uncommon (affects less than 1 in 100 people)
- Too much calcium in your blood (hypercalcaemia)
- Too much calcium in your urine (hypercalciuria)
Rare (affects less than 1 in 1000 people)
- Skin rash
- Itching
- Hives
Reporting of side effects
If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
5. How to store VITANOVA- D3
Keep this medicine out of the sight and reach of children. Do not use this medicine after the expiry date which is stated on the carton and blister pack after "Exp". The expiry date refers to the last day of that month. Do not store above 30° C. Store in the original package in order to protect from light. Do not freeze. Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help to protect the environment.
6. Contents of the pack and other information
What VITANOVA- D3 contains
The active substance is cholecalciferol (vitamin D3).
Each sachet of 1 g contains:
Cholecalciferol IP 60,000 IU
Excipients q.s.
Packaging
4 Sachet of 1g each
For More Information About This Product
Vitanova-D3 SG Capsule
1. Generic Name
Cholecalciferol (Vitamin-D3) Capsules
2. Qualitative and quantitative composition
Each soft gelatin capsules contains:
Cholecalciferol (Vitamin D3) IP 1500 mcg
equivalent to 60,000 IU
Excipients q.s.
Appropriate overages of vitamin added to compensate the loss on storage.
3. Dosage form and strength
Soft Gelatin Capsules
60,000 IU
4. Clinical particulars
4.1 Therapeutic indication
For the treatment of vitamin D3 deficiency
4.2 Posology and method of administration
Adults and adolescent: One Vitanova-D3 SG Capsules (60,000 IU) to be given once a week for a period of 8 weeks, followed by one Vitanova-D3 SG Capsules (60,000 IU) once a month or daily maintenance dose as directed by the physician.
Certain populations are at high risk of vitamin D deficiency, and may require higher doses and monitoring of serum 25(OH)D:
- Institutionalised or hospitalised individuals
- Dark skinned individuals
- Individuals with limited effective sun exposure due to protective clothing or consistent use of sun screens
- Obese individuals
- Patients being evaluated for osteoporosis
- Use of certain concomitant medications (e.g., anticonvulsant medications, glucocorticoids)
- Patients with malabsorption, including inflammatory bowel disease and coeliac disease
- Those recently treated for vitamin D deficiency, and requiring maintenance therapy.
Infants and young children (0-12 years)
Vitanova-D3 SG Capsules should not be given to children under 12 years due to the risk of choking.
Method of administration
This medicine is taken orally.
The Vitanova-D3 SG capsule should be swallowed whole with water, preferably with the main meal of the day.
4.3 Contraindications
- Hypersensitivity to vitamin D or any of the excipients in the product
- Hypervitaminosis D
- Nephrolithiasis
- Diseases or conditions resulting in hypercalcaemia and/or hypercalciuria
- Severe renal impairment
4.4 Special warnings and precautions for use
- Vitamin D should be used with caution in patients with impairment of renal function and the effect on calcium and phosphate levels should be monitored. The risk of soft tissue calcification should be taken into account. In patients with severe renal insufficiency, vitamin D in the form of Cholecalciferol is not metabolised normally and other forms of vitamin D should be used.
- Caution is required in patients receiving treatment for cardiovascular disease.
- Vitanova-D3 should be prescribed with caution to patients suffering from sarcoidosis because of the risk of increased metabolism of vitamin D to its active form. These patients should be monitored with regard to the calcium content in serum and urine.
- Allowances should be made for vitamin D supplements from other sources.
- The need for additional calcium supplementation should be considered for individual patients. Calcium supplements should be given under close medical supervision.
- Medical supervision is required whilst on treatment to prevent hypercalcaemia.
4.5 Drugs interactions
- Concomitant treatment with phenytoin or barbiturates can decrease the effect of vitamin D because of metabolic activation. Concomitant use of glucocorticoids can decrease the effect of vitamin D.
- The effects of digitalis and other cardiac glycosides may be accentuated with the oral administration of calcium combined with Vitamin D. Strict medical supervision is needed and, if necessary monitoring of ECG and calcium.
- Simultaneous treatment with ion exchange resins such as cholestyramine or laxatives such as paraffin oil may reduce the gastrointestinal absorption of vitamin D.
- The cytotoxic agent actinomycin and imidazole antifungal agents interfere with vitamin D activity by inhibiting the conversion of 25-hydroxyvitamin D to 1,25- dihydroxyvitamin D by the kidney enzyme, 25-hydroxyvitamin D-1-hydroxylase.
4.6 Use in special populations
Pregnancy
No data are available for cholecalciferol (vitamin D3). There are no studies in pregnant women. It should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and fetus.
Nursing Mothers
Cholecalciferol and some of its active metabolites pass into breast milk. Infants should be closely monitored for hypercalcemia or clinical manifestations of vitamin D toxicity if the mother is taking pharmacological doses of vitamin D3.
Infants
Vitamin D3 should be used with caution in infants, who may have increased sensitivity to its effects.
4.7 Effects on ability to drive and use machines
Vitanova D3 has no influence on the ability to drive and use machines.
4.8 Undesirable effects
Adverse reactions are listed below, by system organ class and frequency. Frequencies are defined as: uncommon (>1/1,000, <1/100) or rare (>1/10,000, <1/1,000).
Metabolism and nutrition disorders
Uncommon: Hypercalcaemia and hypercalciuria. Skin and subcutaneous disorders
Rare: Pruritus, rash and urticaria.
Reporting of suspected adverse reactions
- Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
- By reporting side effects, you can help provide more information on the safety of this medicine.
4.8 Overdose
The most serious consequence of acute or chronic overdose is hypercalcaemia due to vitamin D toxicity. Symptoms may include nausea, vomiting, polyuria, anorexia, weakness, apathy, thirst and constipation. Chronic overdoses can lead to vascular and organ calcification as a result of hypercalcaemia.
Treatment should consist of stopping all intake of vitamin D and rehydration. Rehydration, and, according to severity, isolated or combined treatment with loop diuretics, bisphosphonates, calcitonin and corticosteroids should be considered. Serum electrolytes, renal function and diuresis must be monitored.
5. Pharmacological properties
5.1 Mechanism of Action
In its biologically active form vitamin D3 stimulates intestinal calcium absorption, incorporation of calcium into the osteoid, and release of calcium from bone tissue. In the small intestine it promotes rapid and delayed calcium uptake. The passive and active transport of phosphate is also stimulated. In the kidney, it inhibits the excretion of calcium and phosphate by promoting tubular resorption. The production of parathyroid hormone (PTH) in the parathyroids is inhibited directly by the biologically active form of vitamin D3. PTH secretion is inhibited additionally by the increased calcium uptake in the small intestine under the influence of biologically active vitamin D3.
5.2 Pharmacodynamic properties
Vitamin D3 is converted to 25-hydroxyvitamin D3 in the liver. Conversion to the active calcium-mobilizing hormone 1, 25-dihydroxyvitamin D3 (calcitriol) in the kidney is stimulated by both parathyroid hormone and hypophosphatemia. The principal action of 1,25-dihydroxyvitamin D3 is to increase intestinal absorption of both calcium and phosphate as well as regulate serum calcium, renal calcium and phosphate excretion, bone formation and bone resorption.
Vitamin D is required for normal bone formation. Vitamin D insufficiency develops when both sunlight exposure and dietary intake are inadequate. Insufficiency is associated with negative calcium balance, increased parathyroid hormone levels, bone loss, and increased risk of skeletal fracture. In severe cases, deficiency results in more severe hyperparathyroidism, hypophosphatemia, proximal muscle weakness, bone pain and osteomalacia.
5.3 Pharmacokinetic properties
The pharmacokinetics of vitamin D is well known. Vitamin D is well absorbed from the gastro-intestinal tract in the presence of bile. It is hydroxylated in the liver to form 25-hydroxyCholecalciferol and then undergoes further hydroxylation in the kidney to form the active metabolite 1, 25 dihydroxy Cholecalciferol (calcitriol). The metabolites circulate in the blood bound to a specific α - globin, Vitamin D and its metabolites are excreted mainly in the bile and faeces.
6. Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Vitamin D is well known and is a widely used material and has been used in clinical practice for many years. As such toxicity is only likely to occur in chronic overdosage where hypercalcaemia could result.
Cholecalciferol has been shown to be teratogenic in high doses in animals (4-15 times the human dose). Offspring from pregnant rabbits treated with high doses of vitamin D had lesions anatomically similar to those of supravalvular aortic stenosis and offspring not showing such changes show vasculotoxicity similar to that of adults following acute vitamin D toxicity.
7. Description
Vitanova D3 contains cholecalciferol (Vitamin D3). Vitamin D3 is essential for the proper growth and development of the body. It is synthesized within the body after exposure to sunlight and is essential for many important functions of the human body. Vitamin D3 in Vitanova D3 also increases the Calcium absorption from the intestines.
8. Pharmaceutical particulars
8.1 Incompatibilities
Not applicable.
8.2 Shelf-life
Refer on the pack
8.3 Packaging information
10 Blister strips of 4 capsules each
8.4 Storage and handing instructions
Store at a temperature not exceeding 30°C. Protect from direct sunlight.
Keep out of reach of children
Capsule should be swallowed whole & not to be opened, chewed or crushed
9. Patient Counselling Information
- Take exactly as directed by your doctor or on the label.
- Do not increase the dosage or take for longer than is recommended.
- Vitanova-D3 Capsules may interfere with cholesterol tests, hence please inform your physician and laboratory staff that you are taking Vitanova-D3 Capsules before undergoing blood tests.
- Clinical monitoring of serum electrolyte concentrations and cardiac function is recommended.
About leaflet
Read all of this leaflet carefully before you start using this medicine, because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist, or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor, or pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet
- What VITANOVA- D3 SG is and what it is used for
- What you need to know before you use VITANOVA- D3 SG
- How to use VITANOVA- D3 SG
- Possible side effects
- How to store VITANOVA- D3 SG
- Contents of the pack and other information
1. What VITANOVA- D3 SG is and what it is used for
VITANOVA- D3 SG capsules contains the active ingredient cholecalciferol (vitamin D3). Vitamin D3, can be found in some foods and is also produced by the body when skin is exposed to sunlight. Vitamin D3 helps the kidneys and intestine absorb calcium and it helps build bones. Vitamin D3 deficiency is the predominant cause of rickets (defective mineralization of bones in children) and osteomalacia (inadequate mineralization of bones in adults).
Certain populations are at high risk of vitamin D deficiency, and may require higher doses and monitoring of serum 25(OH)D:
- Institutionalised, foster homes or hospitalised individuals
- Dark skinned individuals
- Individuals with limited effective sun exposure due to protective clothing or consistent use of sun screens
- Obese individuals
- Patients being evaluated for osteoporosis
- Use of certain concomitant medications (e.g., anticonvulsant medications, glucocorticoids)
- Patients with malabsorption, including inflammatory bowel disease and coeliac disease
- Those recently treated for vitamin D deficiency, and requiring maintenance therapy.
VITANOVA- D3 soft capsules is used for initial treatment of clinically relevant vitamin D deficiency in adults.
2. What you need to know before you use VITANOVA- D3 SG
Do not use VITANOVA- D3:
- if you are allergic to vitamin D3 or any of the other ingredients of this medicine;
- if you have high levels of calcium in your blood (hypercalcaemia) or urine (hypercalciuria);
- if you have kidney stones (renal calculi) or severe renal impairment;
- if you have high levels of vitamin D3 in your blood (hypervitaminosis D)
Warnings and precautions
- Talk to your doctor, pharmacist or nurse before using VITANOVA- D3 if you:
- are undergoing treatment with certain medicines used to treat heart disorders (e.g., cardiac glycosides, such as digoxin);
- have sarcoidosis (an immune system disorder which may cause increased levels of vitamin D3 in the body);
- are treated with diuretics (e.g. benzothiadiazine)
- are immobilized
- suffer from pseudohypoparathyroidism
- are taking medicines containing vitamin D3, or eating foods or milk enriched with vitamin D3;
- are likely to be exposed to a lot of sunshine whilst using VITANOVA- D3;
- take additional supplements containing calcium. Your doctor will monitor your blood levels of calcium to make sure they are not too high whilst you are using VITANOVA- D3;
- have kidney damage or disease and if you have a tendency for the formation of renal stones. Your doctor may want to measure the levels of calcium in your blood or urine. take a daily dose of vitamin D3 exceeding 1,000 I.U. over a long period of time, your doctor should monitor the level of calcium in your blood by lab test.
Children and Adolescents
The use is not recommended in children under 12 years of age.
Other medicines and VITANOVA- D3
Tell your doctor or pharmacist if you are using, have recently used or might use any other medicines. This is especially important if you are taking:
- medicines that act on the heart or kidneys, such as cardiac glycosides (eg, digoxin) or diuretics (e.g., bendroflumethiazide). When used at the same time as vitamin D3 these medicines may cause a large increase in the level of calcium in the blood and urine;
- medicines containing vitamin D3 or eating food rich in vitamin D3, such as, some types of vitamin D3-enriched milk;
- actinomycin (a medicine used to treat some forms of cancer) and imidazole antifungals (eg, clotrimazole and ketoconazole, medicines used to treat fungal disease). These medicines may interfere with the way your body process vitamin D3;
- medicines to treat tuberculosis e.g rifampicin, isoniazid;
- the following medicines because they can interfere with the effect or the absorption of vitamin D3:
- antiepileptic medicines (anticonvulsants), barbiturates;
- glucocorticoids (steroid hormones such as hydrocortisone or prednisolone). These can decrease the effect of vitamin D3;
- medicines that lower the level of cholesterol in the blood (such as cholestyramine, or colestipol);
- certain medicines for weight loss that reduce the amount of fat your body absorbs (eg, orlistat); certain laxatives (such as liquid paraffin).
VITANOVA- D3 SG with food, drink and alcohol
You should take this medicine preferably together with a meal to help your body absorb the vitamin D3.
Pregnancy, breast-feeding and fertility
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine. This high strength formulation is not recommended for use in pregnant and breastfeeding women.
Driving and using machines
VITANOVA- D3 SG should not affect your ability to drive or operate machinery.
3. How to use VITANOVA- D3 SG
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure. This medicine is taken orally.
The capsule should be swallowed whole (e.g. with water), not to be opened, chewed or crushed.
You should take Vitanova-D3 SG preferably together with a meal.
Adults and adolescent: One Vitanova-D3 SG Capsules (60,000 IU) to be given once a week for a period of 8 weeks, followed by one Vitanova-D3 SG Capsules (60,000 IU) once a month or daily maintenance dose as directed by the physician.
Your doctor will adjust the dose for you.
Pediatric population
Vitanova-D3 SG 60, 000 I.U. is not recommended in children under 12 years of age.
Pregnancy and breastfeeding
Vitanova-D3 SG should be used during pregnancy and lactation only if the potential benefit justifies the potential risk to the mother and fetus.
If you are pregnant or think you may be pregnant or you are breast-feeding, you should talk to your doctor or pharmacist before you take Vitanova-D3 SG.
If you take more VITANOVA- D3 SG than you should
If you take more medicine than prescribed, stop using this medicine and contact your doctor. If it is not possible to talk to a doctor go to the nearest hospital emergency department and take the medicine package with you. The most common symptoms of overdose are: nausea, vomiting, excessive thirst, the production of large amounts of urine over 24 hours, constipation and dehydration, high levels of calcium in the blood and in urine (hypercalcaemia and hypercalciuria) shown by lab test.
If you forget to take Vitanova- D3 SG
If you forget to take a dose of Vitanova- D3, take the forgotten dose as soon as possible. Then take the next dose at the correct time. However, if it is almost time to take the next dose, do not take the dose you have missed; just take the next dose as normal. Do not take a double dose to make up for a forgotten dose.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Possible side effects may include:
Uncommon (affects less than 1 in 100 people)
- Too much calcium in your blood (hypercalcaemia)
- Too much calcium in your urine (hypercalciuria)
Rare (affects less than 1 in 1000 people)
- Skin rash
- Itching
- Hives
Reporting of side effects
If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.co.in and click the tab “Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
5. How to store VITANOVA- D3 SG
Keep this medicine out of the sight and reach of children. Do not use this medicine after the expiry date which is stated on the carton and blister pack after "Exp". The expiry date refers to the last day of that month. Do not store above 30° C.
Store in the original package in order to protect from light. Do not freeze. Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help to protect the environment.
6. Contents of the pack and other information
What VITANOVA- D3 contains
The active substance is cholecalciferol (vitamin D3). One capsule contains: 1500 mcg cholecalciferol (vitamin D3) equivalent to 60, 000 IU.
Packaging
10 Blister strips of 4 capsules each