Rifaximax Tablet 550 mg


1.0 Generic name
Rifaximin Tablets IP 200 mg
2.0 Qualitative and quantitative composition
Rifaximax 200
Each film coated tablet contains :
Rifaximin IP 200 mg
Excipients q.s.
Colours : Sunset Yellow FCF & Titanium Dioxide I.P.
3.0 Dosage form and strength
Film-coated tablet.
200 mg
4.0 Clinical particulars
4.1 Therapeutic indication
Rifaximax 200 mg is indicated for For infectious diarrhoea in adults. Rifaximax 400 mg is indicated for For the treatment of hepatic encephalopathy. Rifaximax 550 mg is indicated for Reduction in risk of overt hepatic encephalopathy (HE) recurrence in patients > 18 years of age.
4.2 Posology and method of administration
Posology
Rifaximax 200 mg
200 mg every 8 hours for three days (total 9 doses). Rifaximin must not be used for more than 3 days even if symptoms continue and a second course of treatment must not be taken.
Rifaximax 400 mg
400 mg tablet taken every 8 hours as per the need assessed by treating physician.
Rifaximax 550 mg
Recommended dose : 550 mg twice a day. The clinical benefit was established from a controlled study in which subjects were treated for 6 months. Treatment beyond 6 months should take into consideration the individual balance between benefits and risks, including those associated with the progression of hepatic dysfunction. Rifaximin can be administered with or without food
Paediatric population
The safety and efficacy of Rifaximin tablets in children (aged less than 18 years) have not been established.
Elderly
No dosage adjustment is necessary as the safety and efficacy data of Rifaximin tablets showed no differences between the elderly and the younger patients.
Hepatic impairment
A dosage adjustment for patients with hepatic insufficiency is not necessary.
Renal impairment
Although dosing change is not anticipated, caution should be used in patients with impaired renal function.
Method of administration
Orally with a glass of water
4.3 Contraindications
Rifaximax must not be used in the following conditions :
- Hypersensitivity to rifaximin, rifamycin-derivatives or to any of the excipients.
- Cases of intestinal obstruction.
4.4 Special warnings and precautions for use
Clostridium difficile associated diarrhoea (CDAD) has been reported with use of nearly all antibacterial agents, including rifaximin. The potential association of rifaximin treatment with CDAD and pseudomembranous colitis (PMC) cannot be ruled out.
Due to the lack of data and the potential for severe disruption of gut flora with unknown consequences, concomitant administration of rifaximin with other rifamycins is not recommended.
Patients should be informed that despite the negligible absorption of the drug (less than 1%), like all rifamycin derivatives, rifaximin may cause a reddish discolouration of the urine. Hepatic Impairment : use with caution in patients with severe (Child-Pugh C) hepatic impairment and in patients with MELD (Model for End-Stage Liver Disease) score > 25. Caution should be exercised when concomitant use of rifaximin and a P-glycoprotein such as ciclosporin is needed.
Both decreases and increases in international normalized ratio (in some cases with bleeding events) have been reported in patients maintained on warfarin and prescribed rifaximin. If co-administration is necessary, the international normalized ratio should be carefully monitored with the addition or withdrawal of treatment with rifaximin. Adjustments in the dose of oral anticoagulants may be necessary to maintain the desired level of anticoagulation.
4.5 Interaction with other medicinal products and other forms of interaction
There is no experience regarding administration of rifaximin to subjects who are taking another rifamycin antibacterial agent to treat a systemic bacterial infection. In vitro data show that rifaximin did not inhibit the major cytochrome P-450 (CYP) drug metabolizing enzymes (CYPs1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4). In in vitro induction studies, rifaximin did not induce CYP1A2 and CYP 2B6 but was a weak inducer of CYP3A4.
In healthy subjects, clinical drug interaction studies demonstrated that rifaximin did not significantly affect the pharmacokinetics of CYP3A4 substrates, however, in hepatic impaired patients it cannot be excluded that rifaximin may decrease the exposure of concomitant CYP3A4 substrates administered (e.g. warfarin, antiepileptics, antiarrhythmics, oral contraceptives), due to the higher systemic exposure with respect to healthy subjects.
Both decreases and increases in international normalized ratio have been reported in patients maintained on warfarin and prescribed rifaximin. If co-administration is necessary, the international normalized ratio should be carefully monitored with the addition or withdrawal of rifaximin. Adjustments in the dose of oral anticoagulants may be necessary. An in vitro study suggested that rifaximin is a moderate substrate of P-glycoprotein (P-gp) and metabolized by CYP3A4. It is unknown whether concomitant drugs which inhibit P-gp and/or CYP3A4 can increase the systemic exposure of rifaximin.
In healthy subjects, co-administration of a single dose of ciclosporin (600 mg), a potent Pglycoprotein inhibitor, with a single dose of rifaximin (550 mg) resulted in 83-fold and 124- fold increases in rifaximin mean Cmax and AUC∞. The clinical significance of this increase in systemic exposure is unknown.
The potential for drug-drug interactions to occur at the level of transporter systems has been evaluated in vitro and these studies suggest that a clinical interaction between rifaximin and other compounds that undergo efflux via P-gp and other transport proteins is unlikely (MRP2, MRP4, BCRP and BSEP).
4.6 Special Populations
Pregnancy
There is no or limited data from the use of rifaximin in pregnant women. Animal studies showed transient effects on ossification and skeletal variations in the foetus. As a precautionary measure, use of rifaximin during pregnancy is not recommended.
Breastfeeding
It is unknown whether rifaximin/metabolites are excreted in human milk. A risk to the breast-fed child cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue / abstain from rifaximin therapy taking into account the benefit of breast feeding for the child and the benefit of therapy for the woman.
Fertility
Animal studies do not indicate direct or indirect harmful effects with respect to male and female fertility.
4.7 Effects on ability to drive and use machines
Dizziness has been reported in clinical controlled trials. However, rifaximin has negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
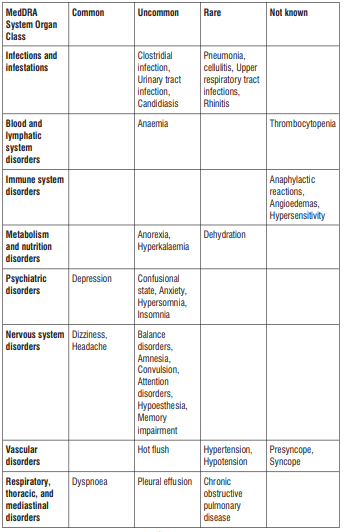
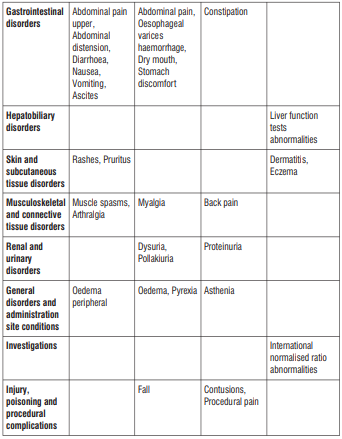
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to : medico@zuventus.com
Website : https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine
4.9 Overdose
No case of overdose has been reported.
In clinical trials with patients suffering from traveller's diarrhoea doses of up to 1800 mg/day have been tolerated without any severe clinical sign. Even in patients/subjects with normal bacterial flora rifaximin in dosages of up to 2400 mg/day for 7 days did not result in any relevant clinical symptoms related to the high dosage.
In case of accidental overdose, symptomatic treatment and supportive care are suggested.
5.0 Pharmacological properties
5.1 Mechanism of action
Rifaximin is an antibacterial drug of the rifamycin class that irreversibly binds the beta sub-unit of the bacterial enzyme DNA-dependent RNA polymerase and consequently inhibits bacterial RNA synthesis.
Rifaximin has a broad antimicrobial spectrum against most of the Gram-positive and negative, aerobic and anaerobic bacteria, including ammonia producing species. Rifaximin may inhibit the division of urea-deaminating bacteria, thereby reducing the production of ammonia and other compounds that are believed to be important to the pathogenesis of hepatic encephalopathy.
5.2 Pharmacodynamic properties
The development of resistance to rifaximin is primarily a reversible chromosomal one-step alteration in the rpoB gene encoding the bacterial RNA polymerase.
Clinical studies that investigated changes in the susceptibility of intestinal flora of patients affected by traveller's diarrhoea failed to detect the emergence of drug resistant Gram-positive (e.g. enterococci) and Gram-negative (E. coli) organisms during a three-day course of treatment with rifaximin.
Development of resistance in the normal intestinal bacterial flora was investigated with repeated, high doses of rifaximin in healthy volunteers and Inflammatory Bowel Disease patients. Strains resistant to rifaximin developed, but were unstable and did not colonise the gastrointestinal tract or replace rifaximin-sensitive strains. When treatment was discontinued resistant strains disappeared rapidly.
Experimental and clinical data suggest that the treatment with rifaximin of patients harbouring strains of Mycobacterium tuberculosis or Neisseria meningitidiswill not select for rifampicin resistance.
Susceptibility
Rifaximin is a non-absorbed antibacterial agent. In vitro susceptibility testing cannot be used to reliably establish susceptibility or resistance of bacteria to rifaximin. There are currently insufficient data available to support the setting of a clinical breakpoint for susceptibility testing.
Rifaximin has been evaluated in vitro on several pathogens including ammonia producing bacteria as Escherichia coli spp, Clostridium spp, Enterobacteriaceae, Bacteroides spp. Due to the very low absorption from the gastro-intestinal tract rifaximin is not clinically effective against invasive pathogens, even though these bacteria are susceptible in vitro
5.3 Pharmacokinetic properties
Absorption
Pharmacokinetic studies in rats, dogs and humans demonstrated that after oral administration rifaximin in the polymorph α form is poorly absorbed (less than 1%). After repeated administration of therapeutic doses of rifaximin in healthy volunteers and patients with damaged intestinal mucosa (Inflammatory Bowel Disease), plasma levels are negligible (less than 10 ng/mL). In HE patients, administration of rifaximin 550 mg twice a day showed mean rifaximin exposure approximately 12-fold higher than that observed in healthy volunteers following the same dosing regimen. A clinically irrelevant increase of rifaximin systemic absorption was observed when administered within 30 minutes of a high-fat breakfast.
Distribution
Rifaximin is moderately bound to human plasma proteins. In vivo, the mean protein binding ratio was 67.5% in healthy subjects and 62% in patients with hepatic impairment when rifaximin 550 mg was administered.
Biotransformation
Analysis of faecal extracts demonstrated that rifaximin is found as the intact molecule, implying that it is neither degraded nor metabolised during its passage through the gastrointestinal tract. In a study using radio-labelled rifaximin, urinary recovery of rifaximin was 0.025% of the administered dose, while <0.01% of the dose was recovered as 25-desacetylrifaximin, the only rifaximin metabolite that has been identified in humans.
Elimination
A study with radio-labelled rifaximin suggested that 14C-rifaximin is almost exclusively and completely excreted in faeces (96.9 % of the administered dose). The urinary recovery of 14Crifaximin does not exceed 0.4% of the administered dose.
6.0 Nonclinical properties
6.1 Animal toxicology or pharmacology
Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenic potential. In a rat embryofetal development study, a slight and transient delay in ossification that did not affect the normal development of the offspring, was observed at 300 mg /kg /day (2.7 times the proposed clinical dose for hepatic encephalopathy, adjusted for body surface area). In the rabbit, following oral administration of rifaximin during gestation, an increase in the incidence of skeletal variations was observed (at doses similar to those proposed clinically for hepatic encephalopathy). The clinical relevance of these findings is unknown.
7.0 Description
Rifaximax tablets contain rifaximin, a non-aminoglycoside semi-synthetic, non-systemic antibiotic derived from rifamycin SV. It is an antibacterial drug Each film-coated tablet of Rifaximax contains Rifaximin 220 / 400 / 550 mg.
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on the pack.
8.3 Packaging information
Rifaximax 200 : A strip of 10 tablets.
Rifaximax 400 : A strip of 10 tablets.
Rifaximax 550 : A strip of 10 tablets.
8.4 Storage and handling instructions
Store in a dry place at a temperature not exceeding 25°C. Protect from light.
Keep out of reach of children.
9.0 Patient counselling information
Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including RIFAXIMAX, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibiotics alters the normal flora of the colon which may lead to C. difficile. Patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If diarrhea occurs after therapy or does not improve or worsens during therapy, advise patients to contact a physician as soon as possible.
Administration with food
Inform patients that Rifaximax may be taken with or without food.
Antibacterial resistance
Counsel patients that antibacterial drugs including RIFAXIMAX should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Rifaximax is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by RIFAXIMAX or other antibacterial drugs in the future.
12.0 Date of revision
12 July 2024

Plot Y2, CTS No : 358/A2, Near Nahur Railway Station,
Nahur (West), Mumbai - 400 078, India.
Registered Trade Mark of Zuventus.
About Leaflet
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor or pharmacist or nurse.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- If you get any side effects, talk to your doctor or pharmacist or nurse. This includes any possible side effects not listed in this leaflet.
What is in this leaflet:
- What RIFAXIMAX® is and what it is used for
- What you need to know before you take RIFAXIMAX ®
- How to take RIFAXIMAX®
- Possible side effects
- How to store RIFAXIMAX®
- Contents of the pack and other information
1. What RIFAXIMAX® is and what it is used for
RIFAXIMAX® is a film-coated tablet that contains the active substance Rifaximin, an intestinal antibiotic. contains the active substance rifaximin. RIFAXIMAX® 200 mg is used for the treatment of infectious diarrhoea in adults, when the diarrhoea is not accompanied by fever or blood in the stools, or 8 or more unformed (soft or liquid) stools in the last 24 hours. RIFAXIMAX® 400 and 550 mg is an antibiotic that destroys bacteria, which can cause a disease called hepatic encephalopathy (symptoms include agitation, confusion, muscle problems, difficulty in speaking and in some cases coma). RIFAXIMAX® 400 mg is used for the treatment of adults with hepatic encephalopathy. RIFAXIMAX® 550 mg is used in adults with liver disease to reduce the recurrence of episodes of overt hepatic encephalopathy.
RIFAXIMAX® 200 mg film-coated tablets are not recommended for use in children (aged less than 18 years). RIFAXIMAX® can either be used alone or more commonly together with medicines containing lactulose (a laxative). You must talk to a doctor if you do not feel better or if you feel worse after 3 days.
2. What you need to know before you take RIFAXIMAX®
- Do not take RIFAXIMAX®:
- If you are allergic (hypersensitive) to rifaximin, to similar types of antibiotics (such as Rifampicin or rifabutin) or to any of the other ingredients of this medicine
- If you have a fever;
- If you have blood in your stools;
- If you passed 8 or more unformed stools in the last 24 hours.
- If you have constipation, abdominal pain and vomiting caused by blockage of the bowel
Warnings and precautions
- Talk to your doctor or pharmacist before taking RIFAXIMAX® film coated tablets and take special care;
- If, after 3 days of treatment, your symptoms continue or re-appear shortly afterwards do not take a second course of RIFAXIMAX® film-coated tablets, see a doctor.
- If your symptoms get worse during treatment stop taking RIFAXIMAX® and consult a doctor.
- While you are taking RIFAXIMAX® your urine may turn a reddish colour. This is quite normal.
- Treatment with any antibiotic including rifaximin may cause severe diarrhoea. This can happen several months after you have finished taking the medicine. If you have severe diarrhoea during or after using RIFAXIMAX® you should stop taking Targaxan and contact your doctor immediately.
Other medicines and RIFAXIMAX®
Please tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines, including medicines obtained without a prescription. Please tell your doctor if you are taking
- Antibiotics (medicines to treat infections),
- Antiepileptics (medicines for the treatment of epilepsy),
- Antiarrhythmics (medicines to treat abnormal heart beat),
- Ciclosporin (a medicine to suppress the body’s immune system),
- Warfarin (medicine to prevent blood clotting), or
- Oral contraceptives (medicines to prevent pregnancy).
If you are using activated charcoal (for example to treat wind or diarrhoea) please take
RIFAXIMAX® film-coated tablets at least 2 hours after taking charcoal.
RIFAXIMAX® with food and drink
RIFAXIMAX® film-coated tablets can be taken with or without food. Orally with a glass of water. Taking this medicine may cause a reddish discolouration of your urine.
Pregnancy and breast-feeding
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking any medicine. It is not known if RIFAXIMAX® can harm your unborn baby. RIFAXIMAX® is therefore not to be used if you are pregnant. It is not known if rifaximin may be passed to your baby in breast milk. RIFAXIMAX® is therefore not to be used if you are breast-feeding.
Inform your doctor
- If you are pregnant, think you may be pregnant or are thinking of becoming pregnant
- If you are breast-feeding or planning to start breast-feeding.
Ask your doctor or pharmacist for advice before taking any medicine.
Driving and using machines
RIFAXIMAX® film-coated tablets are unlikely to affect your reactions when driving or using machines. If you feel dizzy or drowsy you should not drive or operate machinery
3. How to use RIFAXIMAX®
Always take this medicine exactly as your doctor or pharmacist has told you. Check with your doctor or pharmacist if you are not sure.
RIFAXIMAX® 200 mg
Unless otherwise prescribed by the doctor, the usual dose is 1 tablet every 8 hours (600 mg/day). You should continue taking RIFAXIMAX® film-coated tablets for three days even if your symptoms have improved.
Unless otherwise prescribed, the duration of the treatment should not exceed three days. If your symptoms persist for more than three days please see a doctor.
RIFAXIMAX® 400 mg
400 mg tablet is to be taken every 8 hours or as per the need assessed by treating physician.
RIFAXIMAX® 550 mg
550 mg tablet is to be taken every 8 hours or as per the need assessed by treating physician. Rifaximin can be taken with or without food Do not break or crush the tablets.
If you take more RIFAXIMAX® tablets than you should
If you take more than the recommended number of tablets, please contact a doctor.
If you forget to take RIFAXIMAX® tablets
Take the next dose at its normal time. Do not take a double dose to make up for a forgotten tablet.
If you stop taking RIFAXIMAX® tablets
If you do not complete the three days of treatment recommended, your symptoms may worsen.
If you have any further questions on the use of this product, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them. Uncommon (may affect up to 1 in 100 people):
- If you have bleeding from swollen blood vessels in your throat (oesophageal varices).
- If you have severe diarrhoea during or after using this medicine. This may be due to an infection of the intestine.
Not known (frequency cannot be estimated from the available data):
If you get an allergic reaction, hypersensitivity or angioedema. Symptoms include:
- Rash, hives or itchy skin,
- Shortness of breath,
- wheezing Heart racing Swelling of the mouth,
- face,
- ankles or any other part of the body,
- Extreme tiredness, dizziness or feeling faint
If you have any unexpected or unusual bleeding or bruising. This may be due to a decrease in the platelets in your blood which increases the risk of bleeding. Frequency is not known (cannot be estimated from the available data).
Other side effects that may occur
Common (may affect up to 1 in 10 people):
- Depressed mood
- Headache
- Dizziness
- Shortness of breath
- Feeling or being sick
- Accumulation of fluid in the abdominal cavity (ascites)
- Rash or itching Muscle cramps Wind, abdominal bloating, abdominal pain, constipation, diarrhoea, urgency to empty your bowels, nausea, involuntary and painful or ineffective straining, vomiting
- Joint pain
- Swelling of ankles, feet or fingers
Uncommon (may affect up to 1 in 100 people):
Thrush (yeast infections), cold sore, inflammation or infection of the nose and throat
Abnormal test results in white blood cells
Anaemia (reduction in red blood cells which can make the skin pale and cause weakness or breathlessness)
Loss of appetite, loss of body fluid (dehydration)
Abnormal dreams, depressed mood, sleeplessness, nervousness
Numbness, migraine, pins and needles, sinus headache, drowsiness
Double vision
Earache, sensation of the room going round (vertigo)
Increased blood pressure,
Hot flushes
Cough, dry throat, blocked nose, sore throat, runny nose
Upper abdominal pain, indigestion, intestinal movement disorder, dry lips, hard stools, blood in the stools, mucus in stools, taste disorders
Blood test results: increased liver enzyme values
Sunburn
Back pain, muscle cramps, muscle weakness, muscle pain, neck pain
Abnormal urine test results: blood in urine, protein in urine, sugar in urine,
Frequent urination, excessive urination, difficulty or pain passing urine, urinary infection (such as cystitis)
Frequent periods
Fever, chills, cold sweat, increased sweating, flu-like illness, pain.
Hyperkalaemia (high level of potassium in the blood)
Anxiety
Feeling sleepy
Feeling unsteady, falls,
Confusion, loss of or poor memory, loss of concentration, reduced sense of touch, convulsions (fits)
Fluid around the lungs (pleural effusion), Oedema (swelling due to too much fluid in the body)
Abdominal pain
Dry mouth
Rare (may affect up to 1 in 1,000 people):
Chest infections including pneumonia
Cellulitis (inflammation of tissue under skin)
Upper respiratory tract infections (nose, mouth, throat)
Rhinitis (inflammation inside the nose)
Dehydration (body water loss)
Changes in blood pressure
Constant breathing problems (such as chronic bronchitis)
Constipation Back pain
Protein in the urine
Feeling weak Bruising
Pain following surgery
Not known (frequency cannot be estimated from the available data):
Fainting or feeling faint
Skin irritation, eczema (itchy, red, dry skin)
Reduction in platelets (seen in the blood)
Changes in the way the liver is working (seen in blood test)
Changes in blood coagulation (International Normalised Ratio seen in blood test).
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Drug Safety Reporting” located on the top of the home page. By reporting side effects, you can help provide more information on the safety of this medicine. You can also report the side effect with the help of your treating physician.
5. How to store RIFAXIMAX®
Keep this medicine out of the sight and reach of children This medicinal product does not require any special storage conditions. Do not use this medicine after the expiry date which is stated on the label and carton after EXP. The expiry date refers to the last day of that month. Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment. Keep this medicine out of the sight and reach of children.
6. Contents of the pack and other information
RIFAXIMAX® 200
Each film-coated tablet contains:
Rifaximin Ph. Eur. 200 mg
Colours: Sunset Yellow FCF & Titanium Dioxide I.P.
RIFAXIMAX® 400
Each film coated tablet contains:
Rifaximin Ph. Eur. 400 mg
Colours: Sunset Yellow FCF & Titanium Dioxide I.P.
RIFAXIMAX® 550
Each film coated tablet contains:
Rifaximin Ph. Eur. 550 mg
Colours: Sunset Yellow FCF & Titanium Dioxide I.P.














